knitr::opts_chunk$set(cache = TRUE, cache.lazy = FALSE)Introduction
After coalescing colinear alignments, removing translocations of repeat-containing sequences and re-coalescing, colinearity is still broken hundreds of time.
Here we explore the role of inversions in scrambling Oikopleura genomes.
Load R pacakges and data
library('OikScrambling') |> suppressPackageStartupMessages()
load("BreakPoints.Rdata")See
vignette("LoadGenomicBreaks", package = "OikScrambling")
for how the different GBreaks objects are prepared.
Trivial inversions
Documentation
Details can be found in
vignette("GenomicBreaks", package = "GenomicBreaks"), in
vignette("StructuralVariants", package = "GenomicBreaks"),
and ?GenomicBreaks::flagInversions
Number of trivial inversions
More inversions are found after coalescing colinear blocks because of
situations where + - + was + - - + before
collapsing.
The number of detected inversions is further increased after double-coalescing, but not much.
(invs_summary <- data.frame(
align = sapply(gbs, function(gb) sum(flagInversions(gb)$inv)),
mapped = sapply(coa, function(gb) sum(flagInversions(gb)$inv)),
map2 = sapply(coa2, function(gb) sum(flagInversions(gb)$inv))
))## align mapped map2
## Oki_Osa 614 670 704
## Oki_Bar 604 675 705
## Oki_Kum 89 105 122
## Oki_Aom 608 665 695
## Oki_Nor 541 591 611
## Osa_Oki 613 671 706
## Osa_Bar 241 321 330
## Osa_Kum 620 678 708
## Osa_Aom 56 73 79
## Osa_Nor 212 275 279
## Bar_Oki 597 667 695
## Bar_Osa 237 319 327
## Bar_Kum 608 686 711
## Bar_Aom 237 319 335
## Bar_Nor 19 24 27
## Ply_Ros 42 51 55
## Ply_Rob 58 71 76
## Ply_Sav 16 92 92
## Ply_Oki 0 0 0
## Rob_Ros 50 65 71
## Rob_Ply 58 70 77
## Rob_Sav 14 92 92
## Rob_Oki 0 0 0
## Dme_Dbu 697 1003 1003
## Dme_Dsu 20 44 45
## Dme_Dya 13 21 23
## Dme_Dma 12 16 16
## Cni_Cbr 221 300 331
## Cni_Cre 91 212 216
## Cni_Cin 40 132 134
## Cbr_Cni 236 318 358
## Cbr_Cre 92 200 207
## Cbr_Cel 64 166 166
sapply(invs_summary, \(x) tapply(x, row.names(invs_summary) |> OikScrambling:::compDistance(), mean))## align mapped map2
## Cbr_Cel 64.00000 166.00000 166.00000
## Cbr_Cni 236.00000 318.00000 358.00000
## Cbr_Cre 92.00000 200.00000 207.00000
## Cni_Cbr 221.00000 300.00000 331.00000
## Cni_Cin 40.00000 132.00000 134.00000
## Cni_Cre 91.00000 212.00000 216.00000
## Dme_Dbu 697.00000 1003.00000 1003.00000
## Dme_Dma 12.00000 16.00000 16.00000
## Dme_Dsu 20.00000 44.00000 45.00000
## Dme_Dya 13.00000 21.00000 23.00000
## In same pop 54.66667 67.33333 76.00000
## Int – Int 42.00000 51.00000 55.00000
## Int – Rob 55.33333 68.66667 74.66667
## Int/Rob – Oki 0.00000 0.00000 0.00000
## Int/Rob – Sav 15.00000 92.00000 92.00000
## North – North 231.75000 308.50000 317.75000
## Oki – North 600.62500 662.87500 691.87500
sapply(invs_summary, \(x) tapply(x, row.names(invs_summary) |> OikScrambling:::compDistance(), median))## align mapped map2
## Cbr_Cel 64 166.0 166.0
## Cbr_Cni 236 318.0 358.0
## Cbr_Cre 92 200.0 207.0
## Cni_Cbr 221 300.0 331.0
## Cni_Cin 40 132.0 134.0
## Cni_Cre 91 212.0 216.0
## Dme_Dbu 697 1003.0 1003.0
## Dme_Dma 12 16.0 16.0
## Dme_Dsu 20 44.0 45.0
## Dme_Dya 13 21.0 23.0
## In same pop 56 73.0 79.0
## Int – Int 42 51.0 55.0
## Int – Rob 58 70.0 76.0
## Int/Rob – Oki 0 0.0 0.0
## Int/Rob – Sav 15 92.0 92.0
## North – North 237 319.0 328.5
## Oki – North 608 670.5 704.5
sapply(invs_summary, \(x) tapply(x, row.names(invs_summary) |> OikScrambling:::compDistance(), sd))## align mapped map2
## Cbr_Cel NA NA NA
## Cbr_Cni NA NA NA
## Cbr_Cre NA NA NA
## Cni_Cbr NA NA NA
## Cni_Cin NA NA NA
## Cni_Cre NA NA NA
## Dme_Dbu NA NA NA
## Dme_Dma NA NA NA
## Dme_Dsu NA NA NA
## Dme_Dya NA NA NA
## In same pop 35.019042 40.79624 47.57100
## Int – Int NA NA NA
## Int – Rob 4.618802 3.21455 3.21455
## Int/Rob – Oki 0.000000 0.00000 0.00000
## Int/Rob – Sav 1.414214 0.00000 0.00000
## North – North 13.301002 22.35322 26.04323
## Oki – North 25.059572 29.79663 33.17675
invs_summary$pairname <- OikScrambling:::compDistance(rownames(invs_summary))
invs_summary$pairname_s <- OikScrambling:::compDistance(rownames(invs_summary), short = TRUE)
invs_summary$genus <- OikScrambling:::compGenus(rownames(invs_summary))
invs_summary$class <- OikScrambling:::compDistClass(rownames(invs_summary))
invs_summary$target <- sub("_.*", "", rownames(invs_summary))
invs_summary <- invs_summary[invs_summary$class != "Int/Rob – Oki",]
ggplot(invs_summary |> tidyr::pivot_longer(c("align", "mapped", "map2"))) +
aes(value, pairname_s) + geom_point(aes(col = target)) +
ggtitle ("Number of inversions found after different post-processings") +
facet_wrap(~name, ncol= 1)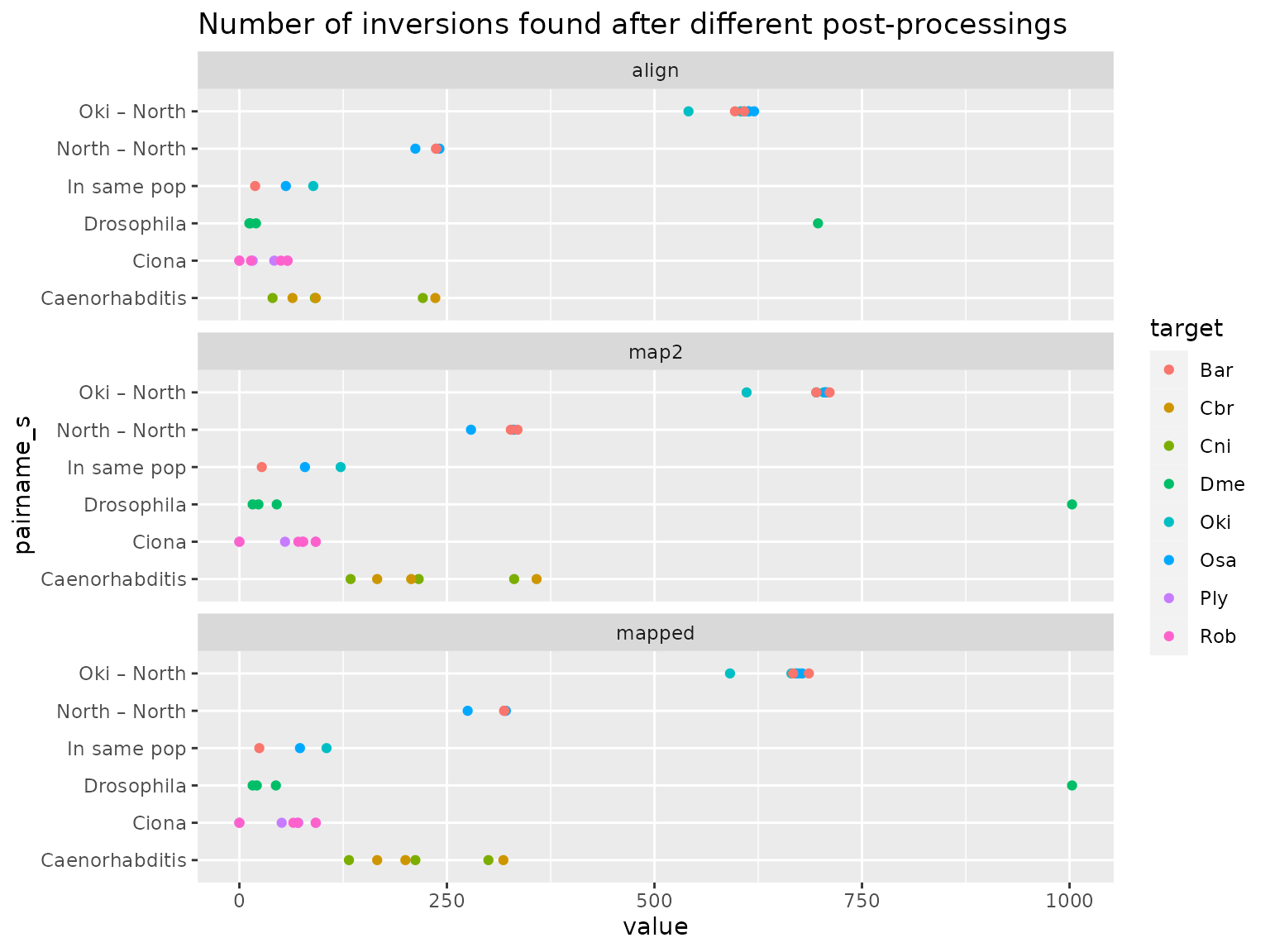
ggplot(invs_summary |> tidyr::pivot_longer(c("mapped"))) +
aes(value, genus) + geom_point(aes(col = class)) +
ggtitle ("Number of inversions found in mapped regions") 
Motif search
Enrichment for motifs is being searched in
vignette("PWM", package = "OikScrambling").
Breakpoint regions flanking inversions in the same-population alignments.
Check
vignette("ColinearityInterruptors", package = "OikScrambling").
Breakpoint regions flanking large inversions in the North–North alignments.
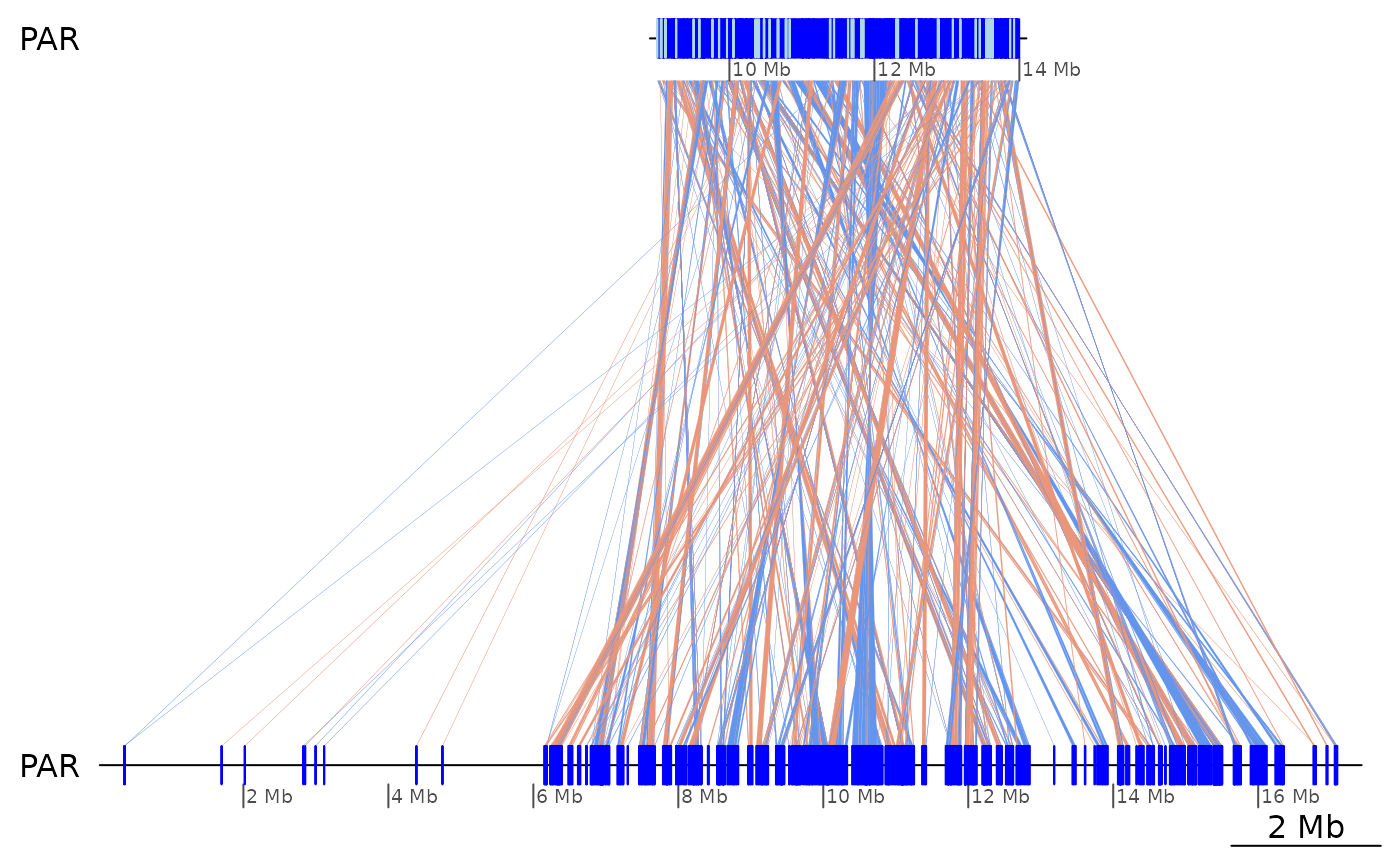
Let’s try to isolate some inversions that are easy to spot on a whole-chromosome alignment.
plotApairOfChrs(coa2$Osa_Bar, "PAR")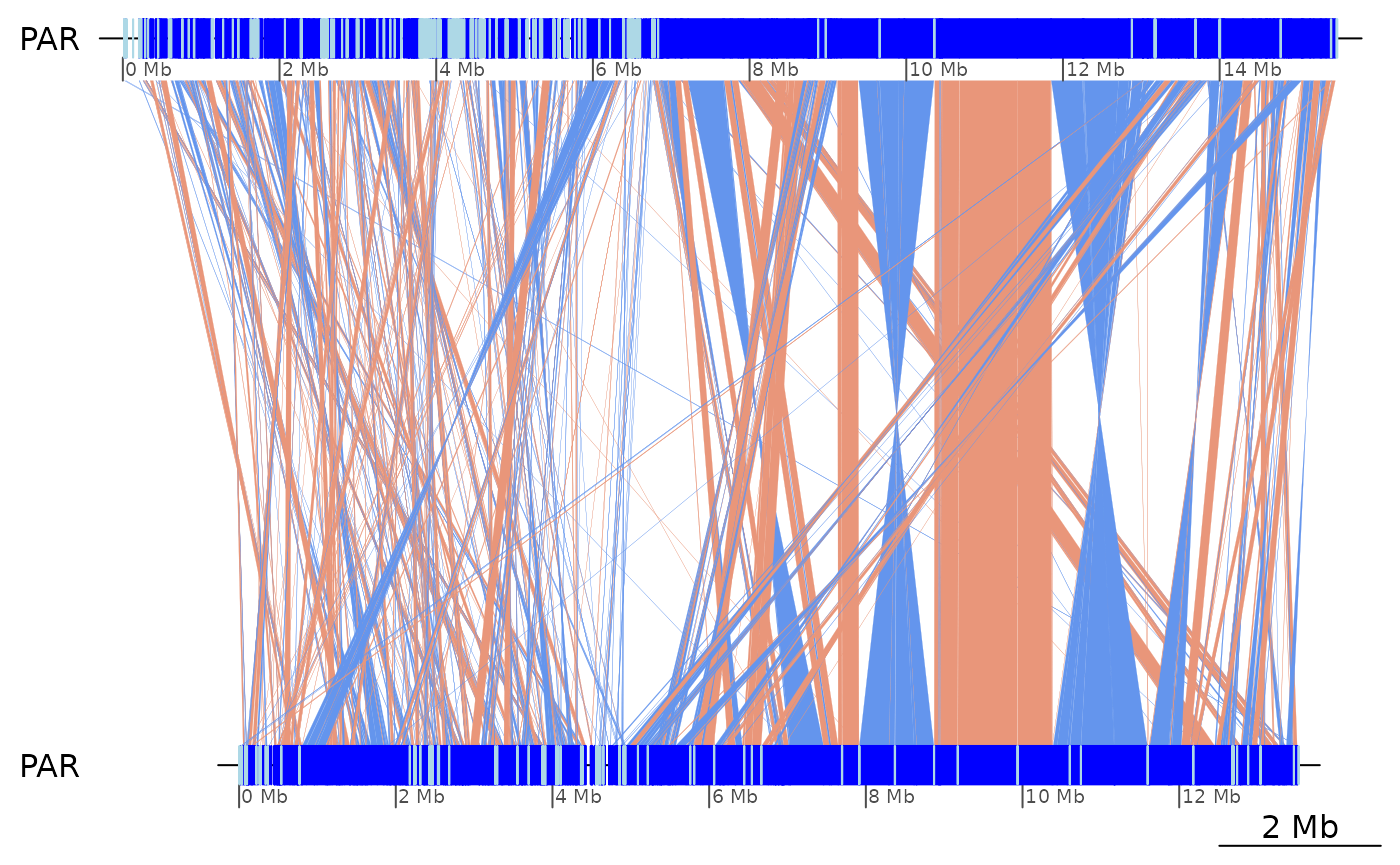
(ROI1 <- coa2$Osa_Bar |> plyranges::filter(seqnames == "PAR", start > 9e6, end < 14e6, start(query) > 7e6, end(query) < 12e6))## GBreaks object with 35 ranges and 6 metadata columns:
## seqnames ranges strand | query Arm
## <Rle> <IRanges> <Rle> | <GRanges> <factor>
## [1] PAR 9112106-9113024 + | PAR:7914713-7915633 long
## [2] PAR 9122674-9169015 + | PAR:7647348-7694603 long
## [3] PAR 9169027-9383526 + | PAR:7700204-7911308 long
## [4] PAR 9393845-9625372 - | PAR:8648572-8868624 long
## [5] PAR 9625425-9626193 + | PAR:8647713-8648486 long
## ... ... ... ... . ... ...
## [31] PAR 12882838-12884857 + | PAR:11615009-11617029 long
## [32] PAR 12884875-13019548 - | PAR:10442170-10570866 long
## [33] PAR 13027569-13077236 - | PAR:10392248-10442013 long
## [34] PAR 13077254-13079729 + | PAR:11592137-11594612 long
## [35] PAR 13079758-13079924 + | PAR:10378144-10378306 long
## rep repOvlp transcripts
## <CharacterList> <integer> <Rle>
## [1] <NA> 0 <NA>
## [2] rnd,tandem 1671 g9531.t1;g9532.t1;g9..
## [3] rnd,unknown,tandem,... 4642 g9552.t1;g9553.t1;g9..
## [4] rnd,tandem,LowComplexity,... 5474 g9629.t1;g9630.t1;g9..
## [5] <NA> 0 <NA>
## ... ... ... ...
## [31] rnd 1167 g10776.t1
## [32] tandem,rnd,LowComplexity,... 3102 g10777.t1;g10779.t1;..
## [33] unknown,rnd,tandem 1637 g10840.t1;g10841.t1;..
## [34] rnd 0 <NA>
## [35] <NA> 0 <NA>
## flag
## <character>
## [1] <NA>
## [2] <NA>
## [3] <NA>
## [4] Inv
## [5] <NA>
## ... ...
## [31] <NA>
## [32] <NA>
## [33] <NA>
## [34] <NA>
## [35] <NA>
## -------
## seqinfo: 483 sequences from OSKA2016v1.9 genome
plotApairOfChrs(ROI1)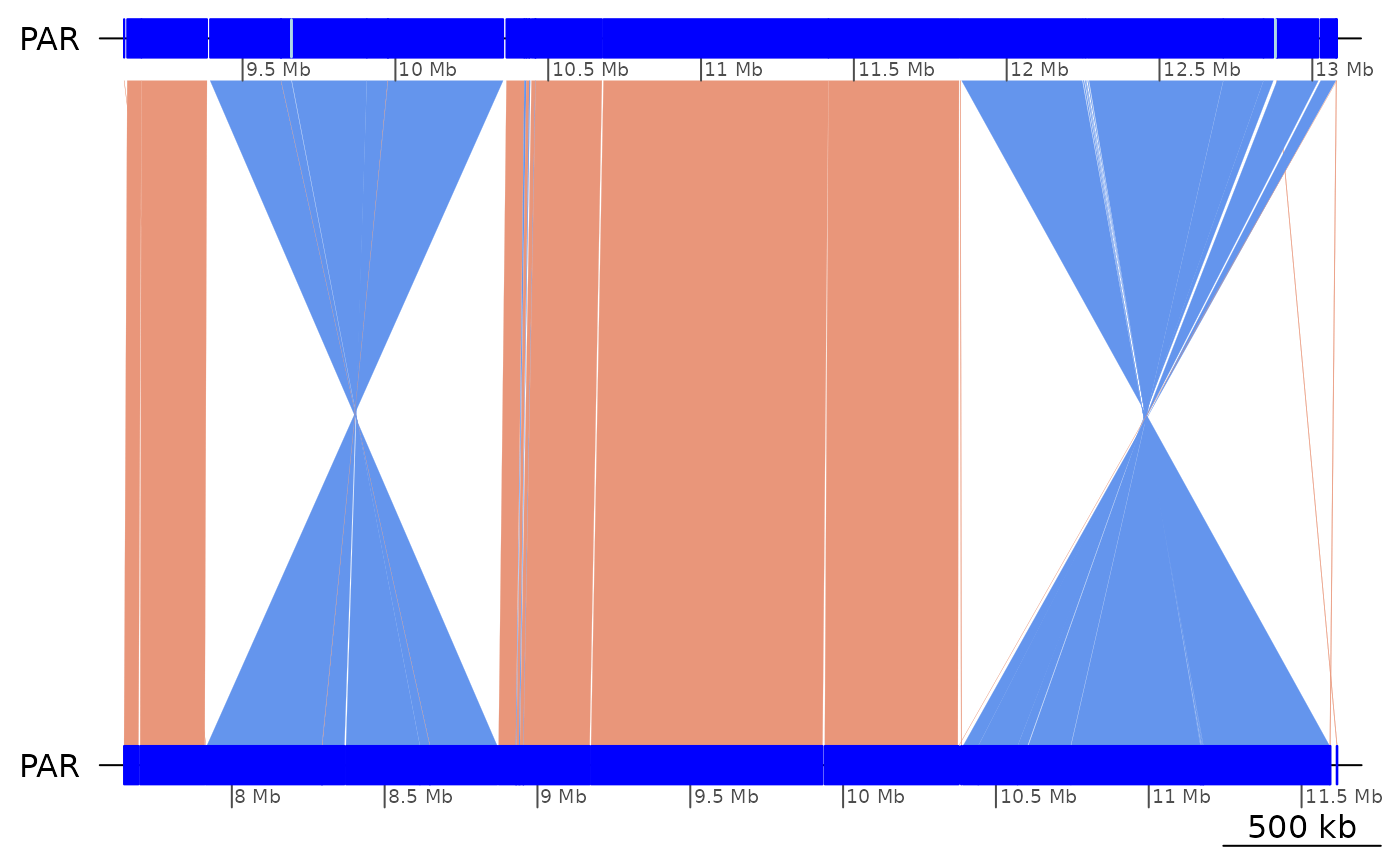
# The contours of the main 4 blocks can be coalesced by applying a size threshold.
ROI1 |> coalesce_contigs(min = 1e4)## GBreaks object with 4 ranges and 2 metadata columns:
## seqnames ranges strand | query score
## <Rle> <IRanges> <Rle> | <GRanges> <integer>
## [1] PAR 9122674-9383526 + | PAR:7647348-7911308 260853
## [2] PAR 9393845-10352419 - | PAR:7917928-8868624 958575
## [3] PAR 10363392-11843885 + | PAR:8872985-10375361 1480494
## [4] PAR 11852584-13077236 - | PAR:10392248-11592118 1224653
## -------
## seqinfo: 483 sequences from OSKA2016v1.9 genome
# The gaps flanking the first inverted region are quite large!
(ROI1_gaps <- ROI1 |> coalesce_contigs(min = 1e4) |> cleanGaps() |> plyranges::mutate(w = width))## GRanges object with 3 ranges and 1 metadata column:
## seqnames ranges strand | w
## <Rle> <IRanges> <Rle> | <integer>
## [1] PAR 9383527-9393844 * | 10318
## [2] PAR 10352420-10363391 * | 10972
## [3] PAR 11843886-11852583 * | 8698
## -------
## seqinfo: 483 sequences from OSKA2016v1.9 genome
# The left-side gap contains no alignment at all even before discarding the short ones.
subsetByOverlaps(gbs$Osa_Bar, ROI1_gaps)## GBreaks object with 4 ranges and 8 metadata columns:
## seqnames ranges strand | score query
## <Rle> <IRanges> <Rle> | <numeric> <GRanges>
## [1] PAR 10355613-10355774 - | 500 XSR:671520-671674
## [2] PAR 10355951-10356573 - | 1610 XSR:670597-671182
## [3] PAR 10356752-10361833 - | 13551 XSR:665505-670596
## [4] PAR 11847299-11849269 + | 5701 PAR:10385920-10388171
## Arm rep repOvlp transcripts flag nonCoa
## <factor> <CharacterList> <integer> <Rle> <character> <logical>
## [1] long <NA> 0 <NA> Col FALSE
## [2] long <NA> 0 <NA> Col FALSE
## [3] long <NA> 0 <NA> <NA> FALSE
## [4] long <NA> 0 g10434.t1 <NA> TRUE
## -------
## seqinfo: 483 sequences from OSKA2016v1.9 genome
# The same region aligns well to Aomori, suggesting that it is not missassembled
coa2$Osa_Aom |> plyranges::filter(seqnames == "PAR", start > 9e6, end < 14e6) |> plotApairOfChrs()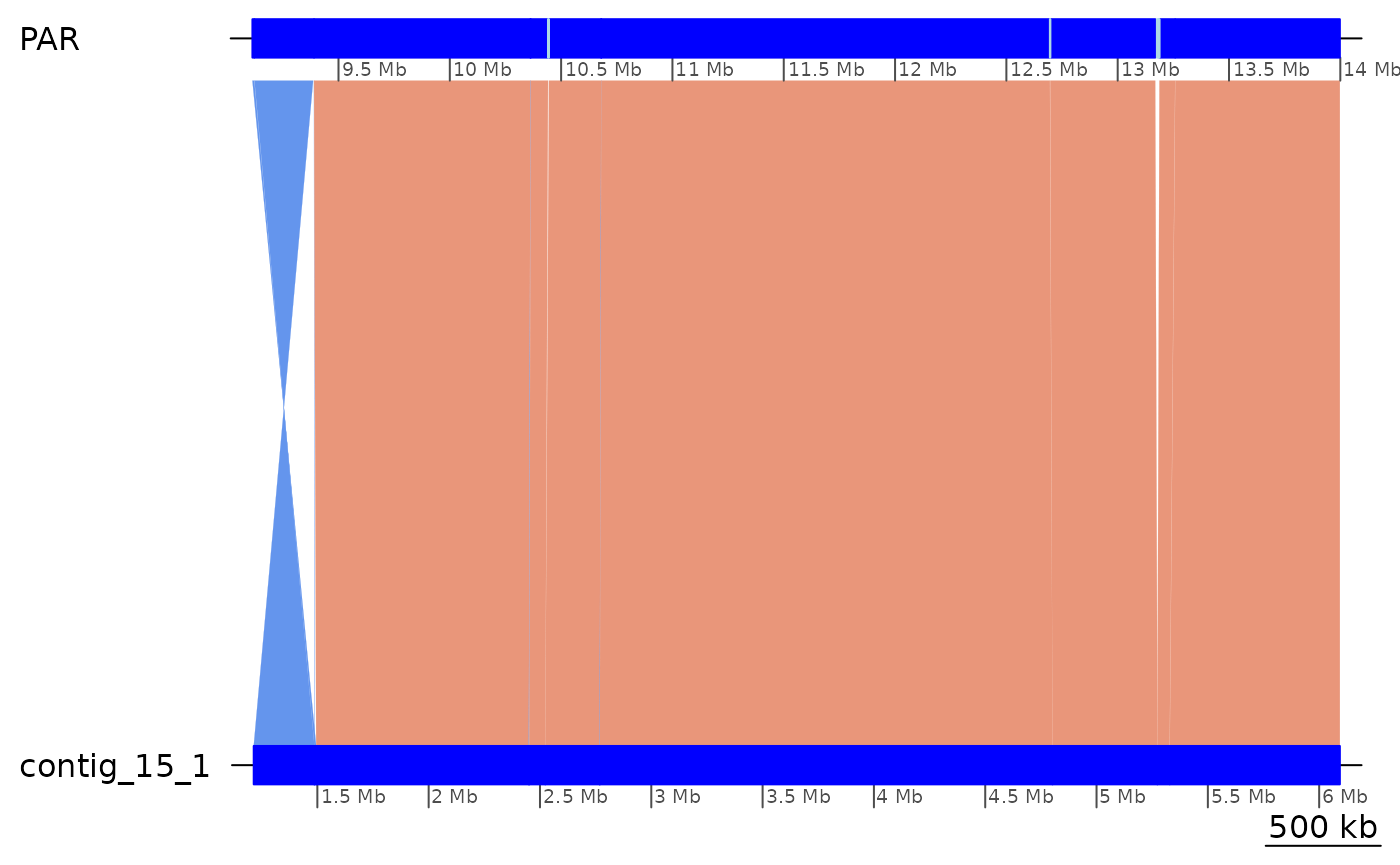
# The Osa–Bar unaligned regions appear to align well to Aom, but the first one is split
# and the second one contains a small inversion. Are there a hotspots ?
subsetByOverlaps(gbs$Osa_Aom, ROI1_gaps, type = "any") |> plotApairOfChrs()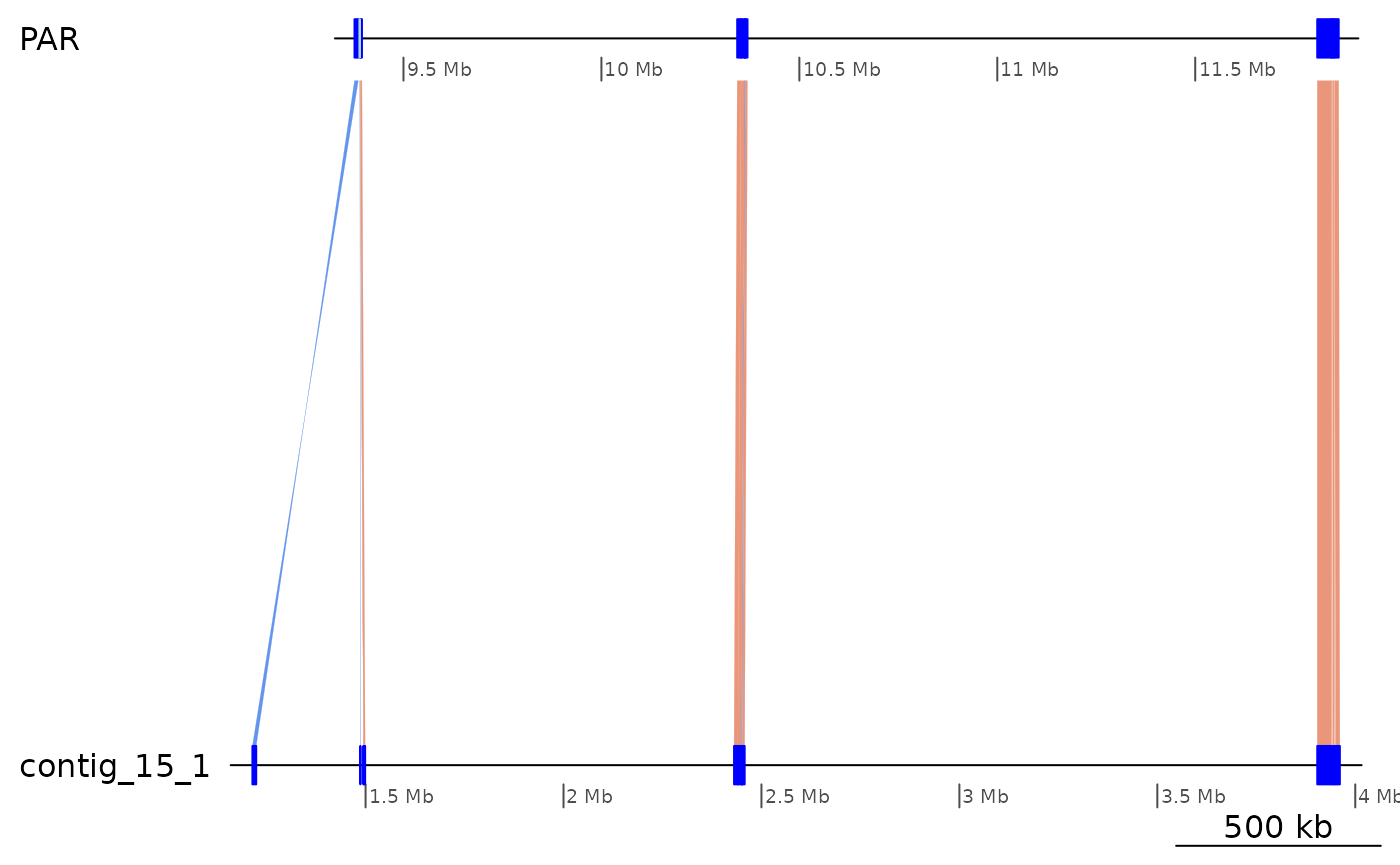
subsetByOverlaps(gbs$Osa_Aom, ROI1_gaps[1], type = "within") |> plotApairOfChrs()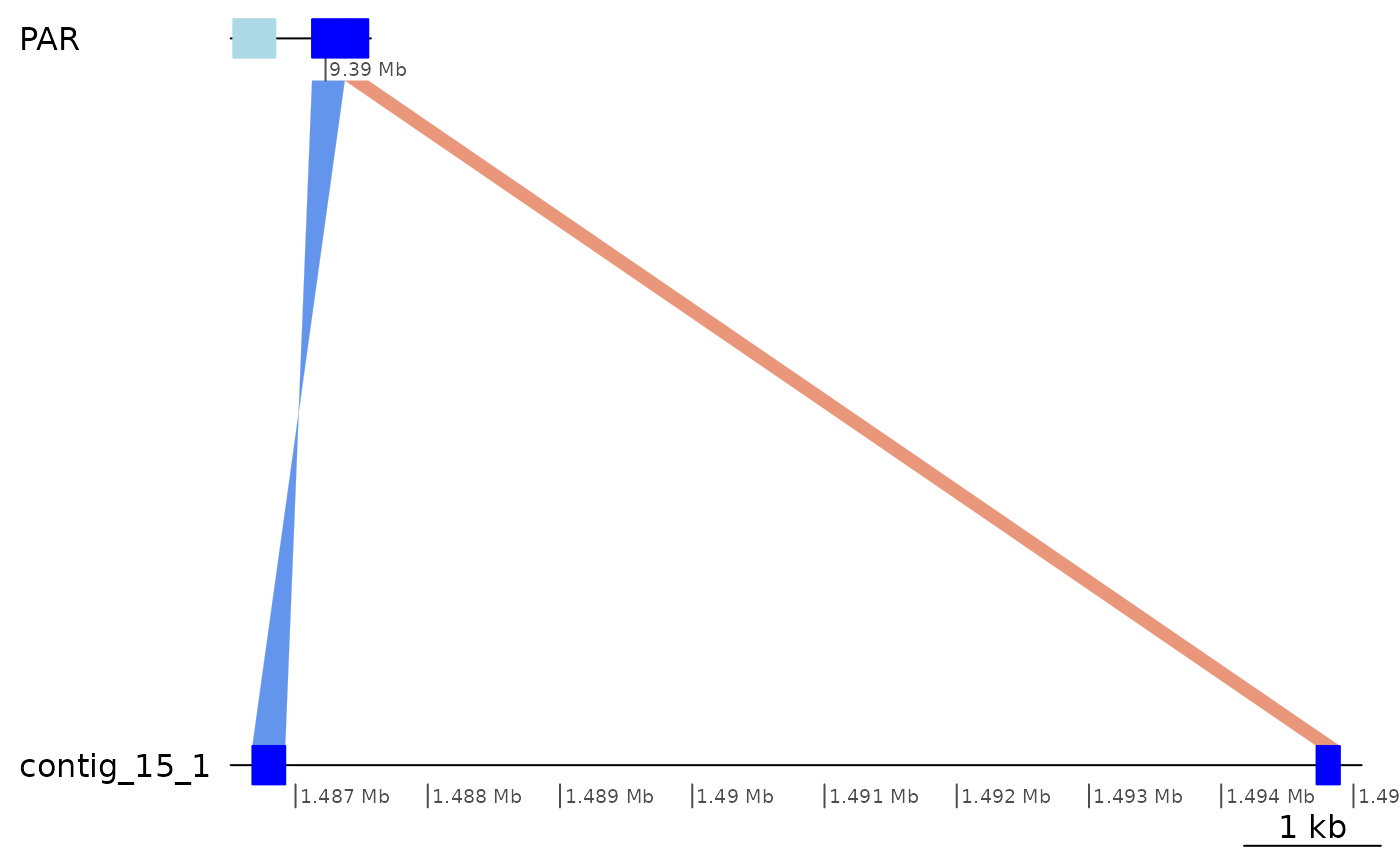
subsetByOverlaps(gbs$Osa_Aom, ROI1_gaps[2], type = "within") |> plotApairOfChrs()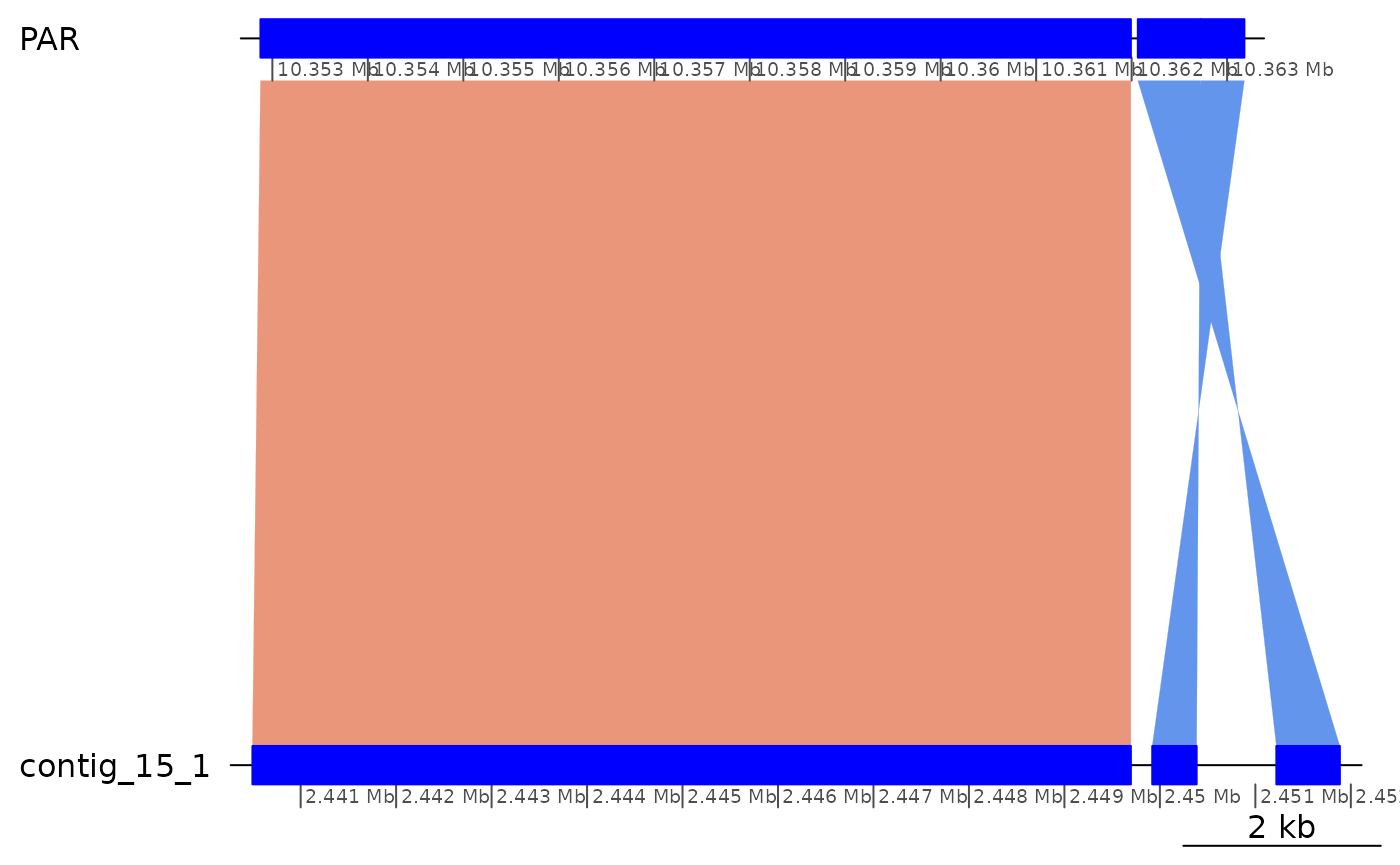
subsetByOverlaps(gbs$Osa_Aom, ROI1_gaps[3], type = "within") |> plotApairOfChrs()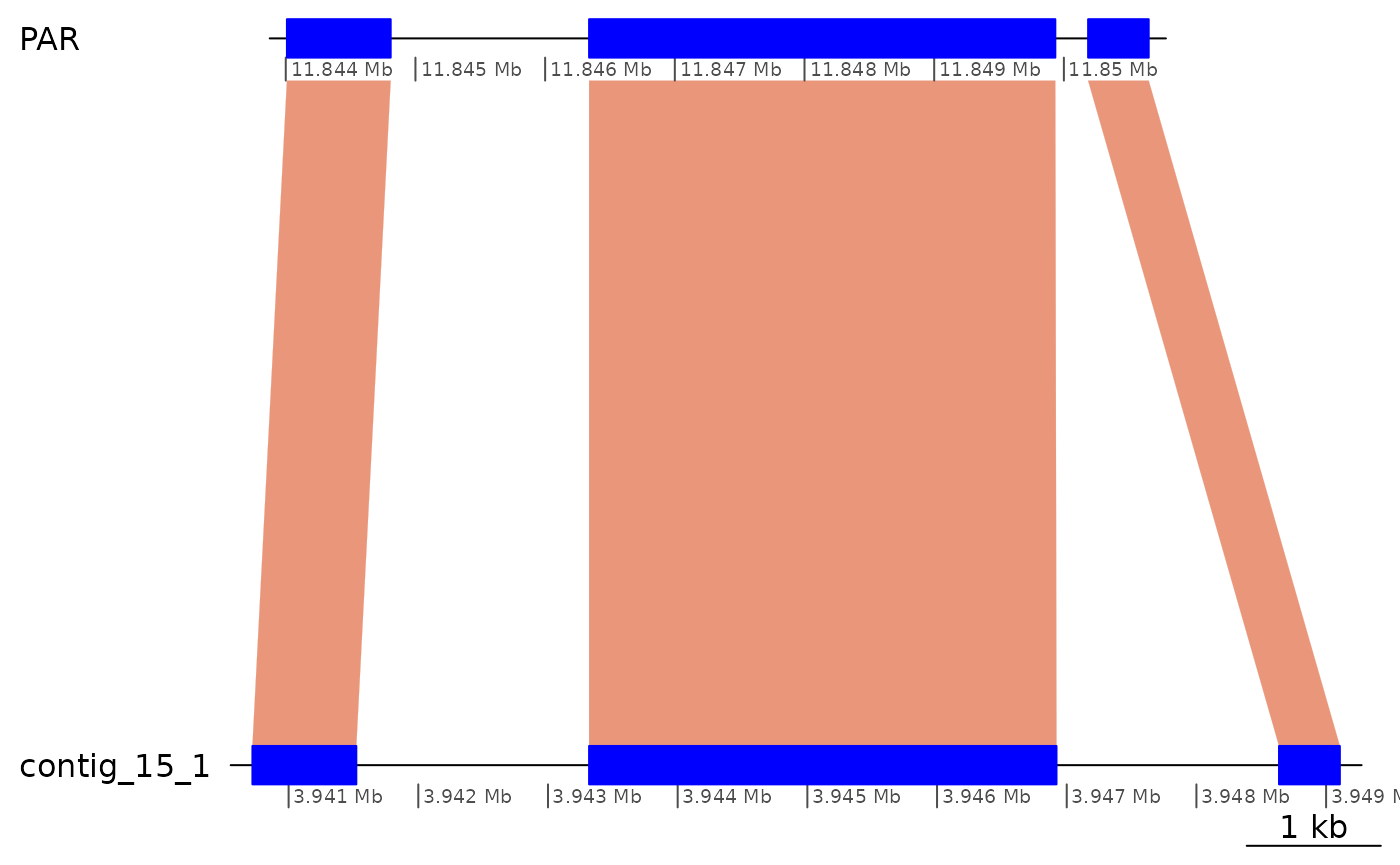
# In the alignment to Okinawa, it is scrambled.
coa2$Osa_Oki |> plyranges::filter(seqnames == "PAR", start > 9e6, end < 14e6) |> plotApairOfChrs()