Genomic features of the breakpoints
Charlotte West
Charles Plessy
20 November, 2023
Source:vignettes/GenomicFeatures.Rmd
GenomicFeatures.RmdIntroduction
Breakpoint feature analyses.
Figure 3 panel C is produced by this vignette.
It also computes operons as we need their boundaries in the figure panel.
** Figure 4 panel C is produced by this vignette**
The vignette also contains extra material that is not entirely
relevant to the manuscript, but that can not be moved elsewhere because
it needs the orthogroup GBreaks object generated at the
beginning at the top.
Load packages and data
See ?OikScrambling:::loadAllGenomes,
?OikScrambling:::loadAllTranscriptsGR, and
vignette("LoadGenomicBreaks", package = "OikScrambling")
for how the different objects are prepared.
library('OikScrambling') |> suppressPackageStartupMessages()
library('GenomicFeatures') |> suppressPackageStartupMessages()
library('heatmaps') |> suppressPackageStartupMessages()
library('patchwork') |> suppressPackageStartupMessages()
genomes <- OikScrambling:::loadAllGenomes(compat = F)## Warning in runHook(".onLoad", env, package.lib, package): input string
## 'Génoscope' cannot be translated from 'ANSI_X3.4-1968' to UTF-8, but is valid
## UTF-8
## Warning in runHook(".onLoad", env, package.lib, package): input string
## 'Génoscope' cannot be translated from 'ANSI_X3.4-1968' to UTF-8, but is valid
## UTF-8
# Cannot use Knitr cache as long as "annot" objects are used.
annots <- OikScrambling:::loadAllAnnotations() |> suppressWarnings()
reps <- OikScrambling:::loadAllRepeats(compat = F)
genes <- sapply(annots, function(a) sort(genes(a))) |> SimpleList()
load("BreakPoints.Rdata")
requireNamespace("rGADEM") |> suppressPackageStartupMessages()
requireNamespace("ggseqlogo") |> suppressPackageStartupMessages()
ces <- readRDS("CEs.Rds")
tfbs <- readRDS("pwmMatchesOki_12_95.Rds")Compute operons
See the
vignette("OperonDetection", package = "OikScrambling") for
details.
operons <- lapply(annots, \(a) {a |> genes() |> OikScrambling:::calcOperons(window = 500) }) |> SimpleList() |> suppressWarnings()
operons$Nor <- OikScrambling:::calcOperons(window = 500, transcripts(annots$Nor) |> reduce()) |> suppressWarnings()
sapply(operons, length)## Oki Osa Bar Kum Aom Nor Ply Ros
## 3124 2741 2379 2974 2648 2996 1088 1472## $Oki
##
## 2 3 4 5 6 7 8 9 10 11 14
## 1943 647 259 134 69 33 19 10 6 3 1
##
## $Osa
##
## 2 3 4 5 6 7 8 9 10 11 12 13
## 1714 557 248 98 63 32 13 10 1 3 1 1
##
## $Bar
##
## 2 3 4 5 6 7 8 9 10 11 12 13 14 17
## 1450 475 221 106 57 30 23 9 3 1 1 1 1 1
##
## $Kum
##
## 2 3 4 5 6 7 8 9 10 11 12 13 14 17
## 1855 648 233 114 56 28 18 4 7 6 1 1 2 1
##
## $Aom
##
## 2 3 4 5 6 7 8 9 10 11 12 14
## 1689 492 239 107 52 36 14 10 3 4 1 1
##
## $Nor
##
## 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17
## 1724 562 264 138 93 65 38 40 16 21 11 10 3 4 3 1
## 20 22
## 1 2
##
## $Ply
##
## 2 3 4 5 6 7 8 9 13 14 15
## 937 114 22 6 2 1 1 1 1 1 2
##
## $Ros
##
## 2 3 4 5 6 7 9 10 11 21 26 29
## 1262 163 33 5 1 1 1 1 1 1 1 2Gene feature coverage around aligned and unaligned genomic regions.
Accessory functions
prepareFreqPlot <- function(gb, annot = NULL, rep = NULL, cne = NULL, tfbs = NULL, operons = NULL, win = 1000, direction = "left", withGenes=c("yes", "no", "only")) {
withGenes <- match.arg(withGenes)
l <- list()
# Note that get_bps(direction="mid") is probably not what you think.
if(direction == "mid") {
gb <- resize(gb, 1, "center")
direction <- "left"
}
if (!is.null(annot)) {
if (withGenes %in% c("yes", "only")) {
l$genes <- feature_coverage(gb, genes(annot), win = win, lab = "genes", direction = direction)
}
if (withGenes %in% c("yes", "no")) {
l$intron <- feature_coverage(gb, intronicParts(annot), win = win, lab = "intron", direction = direction)
l$exon <- feature_coverage(gb, exonicParts(annot), win = win, lab = "exon", direction = direction)
#reorder
newOrder <- order(rowSums(l$intron@image))
l$intron@image <- l$intron@image[newOrder,]
l$exon@image <- l$exon@image[newOrder,]
}
}
if (!is.null(operons))
l$operons<-feature_coverage(gb, operons, win = win, lab = "operons",direction = direction)
if (!is.null(rep))
l$reps <- feature_coverage(gb, rep, win = win, lab = "repeats",direction = direction)
if (!is.null(cne))
l$CNE <- feature_coverage(gb, cne, win = win, lab = "CNE", direction = direction)
if (!is.null(tfbs))
l$tfbs <- feature_coverage(gb, tfbs, win = win, lab = "TFBS", direction = direction)
l
}
#' Prepare a long tibble for frequency plotting
#'
#' Loop over a list of `Heatmap` objects to produce a long tibble that can be
#' used with ggplot to plot frequency of each feature included in the list.
#'
#' @param x A list of `Heatmap` objects
#' @param desc An additional description (character string)
#'
#' @return A long `tibble` that is the vertical concatenation of all tibbles
#' produced for each element of the list `x`. Its `what` column is made from
#' the names of the heatmaps in the list, and its `desc` column is made from the
#' `desc` arguments. The other columns (`val`, `freq` and `pos`) originate from
#' the `Heatmap_to_freq_tibble` function call.
prepareGGFreqPlot <- function(x, desc = NULL) {
l <- lapply(names(x), \(name){
tib <- Heatmap_to_freq_tibble(x[[name]], desc = desc)
tib$what <- name
tib
})
do.call(rbind, l)
}
#' Heatmap to long tibble for ggplot
#'
#' Coerce a Heatmap object into a \dQuote{long} tibble with one value per
#' position, either as raw counts or as a frequency.
#'
#' The tibble can then be passed to ggplot. The `image` matrix of the `Heatmap`
#' object has one column per nucleotide position and it is assumed that its
#' central column is for position zero.
#'
#' @param h a `Heatmap` object
#'
#' @return a `tibble` with `val`, `freq` and `pos` columns
Heatmap_to_freq_tibble <- function(h, desc = NULL) {
val <- colSums(h@image)
tibble::tibble( val = val
, freq = val / nrow(h@image)
, pos = 1:ncol(h@image) - round(ncol(h@image)) / 2
, desc = desc)
}Centering on left-aligned boundaries.
Breakpoints are between mapped regions. If the unmapped region is broad we have little information where the breakpoint originally was. We set arbitrarly the breakpoints to the left-side (end-side) coordinate of genomic regions.
Mapped regions can consist of multiple colinear aligned regions, or be a single uncoalesced aligned region. We will see that they have different properties.
Our annotations is mostly made of coding exons, so the features exons and cds have very similar profiles. Therefore we keep only exons in the rest of the analysis.
Positive coordinates (and zero) start in selected regions and negative coordinates start outside these regions. As we move further from 0, we can not guarantee that the statement is still true because some regions have a width narrower than the plot window. This is why the values tend to equilibrate away from the boundaries.
Aligned regions are enriched in exons and depleted in introns.
Close to the breakpoint of uncoalesced mapped regions, exon annotations are depleted, which makes sense, as exons are usually mapable. Within the mapped regions, frequency of introns and exons are comparable. In contrary, within coalesced mapped regions, the exon annotations are over-represented.
#' Pre-compute feature frequencies in fixed windows
#'
#' @param target 3-letter name of one Oik genome
#' @param query 3-letter name of another Oik genome
#' @param perpareFreqPlot a `perpareFreqPlot` function
#' @param reps `SimpleList` of `GRanges` of repeat regions
#' @param CE `SimpleList` of `GRanges` with conserved non-coding elements
#' @param tfbs `SimpleList` of `GRanges` of putative transcription factor binding sites
#' @param operons `SimpleList` of `GRanges` of operon regions.
#' @param direction Where to center the windows (`left`, `right` or `mid`)
#'
#' @return A `SimpleList` of Heatmap objects
computePlotData <- function(target, query, annots=NULL, reps=NULL, CE=NULL, tfbs=NULL, operons=NULL, direction="left") {
pair <- paste(target, query, sep = "_")
current_GB_aln_nonCoa <- gbs[[pair]][ gbs[[pair]]$nonCoa]
current_GB_aln_coa <- gbs[[pair]][ ! gbs[[pair]]$nonCoa]
current_GB_syn_block <- coa[[pair]][ ! coa[[pair]]$nonCoa]
# We do not use unal, because it overlaps with bri
current_GB_unmap <- unmap[[pair]]
current_GB_bri <- bri[[pair]]
targetGenome <- unique(genome(gbs[[pair]]))
prepareFreqPlot_default <- \(x) prepareFreqPlot(x, annot = annots[[target]], rep = reps[[targetGenome]], cne = CE[[pair]], tfbs = tfbs, operons = operons[[target]], direction = direction)
hmList <- SimpleList()
hmList$aln_nonCoa <- prepareFreqPlot_default(current_GB_aln_nonCoa)
hmList$aln_coa <- prepareFreqPlot_default(current_GB_aln_coa)
hmList$syn_block <- prepareFreqPlot_default(current_GB_syn_block)
hmList$unmap <- prepareFreqPlot_default(current_GB_unmap)
hmList$bri <- prepareFreqPlot_default(current_GB_bri)
hmList
}
hmList_to_plot_noncoding <- computePlotData("Oki", "Osa", reps = reps, CE = ces$ce__50__48)
hmList_to_plot_coding <- computePlotData("Oki", "Osa", annots = annots, operons = operons)
list_of_heatmaps_to_long_tibble <- function(hmList_to_plot, desc = NULL) {
tib <-rbind(
hmList_to_plot$aln_nonCoa |> prepareGGFreqPlot("01_isolated_alns"),
hmList_to_plot$unmap |> prepareGGFreqPlot("02_unmapped"),
hmList_to_plot$syn_block |> prepareGGFreqPlot("03_colinear_reg"),
hmList_to_plot$bri |> prepareGGFreqPlot("04_bridge"),
hmList_to_plot$aln_coa |> prepareGGFreqPlot("05_colinear_alns")
)
tib$desc2 <- desc
tib
}
rbind(
list_of_heatmaps_to_long_tibble(hmList_to_plot_noncoding, "noncoding"),
list_of_heatmaps_to_long_tibble(hmList_to_plot_coding, "coding")
) |> ggplot() +
aes(pos, freq, col = what) +
geom_line() +
facet_wrap(~desc2 + desc, nrow = 2) +
theme_bw() +
ggtitle("Aligned on regions left boundary. CNE is 50/48. Okinawa - Osaka")
Most of the narrow-width aligned regions did not coalesce. We use
this as a classifier instead of an arbitrary width cutoff. See
vignette("RegionWidths", package = "OikScrambling") for
details.
Figure 3C panel
,With only 2 panels, for unmapped–aligned and bridge–aligned, we display almost all the information present in the 5-panel version above.
computePlotData_simplified <- function(target, query, annots=NULL, reps=NULL, CE=NULL, tfbs=NULL, operons=NULL, direction="left", withGenes = c("yes", "no", "only")) {
withGenes <- match.arg(withGenes)
pair <- paste(target, query, sep = "_")
current_GB_brk <- unmap[[pair]] |> subsetByOverlaps(coa[[pair]][!coa[[pair]]$nonCoa] + 1) # The unmapped region flanking colinear regions
current_GB_bri <- bri[[pair]]
targetGenome <- unique(genome(gbs[[pair]]))
prepareFreqPlot_default <- \(x) prepareFreqPlot(x, annot = annots[[target]], rep = reps[[targetGenome]], cne = CE[[pair]], tfbs = tfbs, operons = operons[[target]], direction = direction, withGenes = withGenes)
hmList <- SimpleList()
hmList$brk <- prepareFreqPlot_default(current_GB_brk)
hmList$bri <- prepareFreqPlot_default(current_GB_bri)
hmList
}
list_of_heatmaps_to_long_tibble <- function(hmList_to_plot, desc = NULL) {
tib <-rbind(
hmList_to_plot$brk |> prepareGGFreqPlot("01_break"),
hmList_to_plot$bri |> prepareGGFreqPlot("02_bridge")
)
tib$desc2 <- desc
tib
}
twoBySixFigure <- function(noncod, genesop, exonintron)
rbind(
list_of_heatmaps_to_long_tibble(noncod, "noncoding"),
list_of_heatmaps_to_long_tibble(genesop, "genes and operons"),
list_of_heatmaps_to_long_tibble(exonintron, "exons and introns")
) |> ggplot() +
aes(pos, freq, col = what) +
geom_line() +
facet_wrap(~desc2 + desc, nrow = 3) +
theme_bw() Okinawa–Osaka
pOkiOsa <- twoBySixFigure(
computePlotData_simplified("Oki", "Osa", reps = reps, CE = ces$ce__50__48, direction = "right"),
computePlotData_simplified("Oki", "Osa", annots = annots, operons = operons, direction = "right", withGenes = "only"),
computePlotData_simplified("Oki", "Osa", annots = annots, direction = "right", withGenes = "no")
) +
ggtitle("Okinawa - Osaka")
pOkiOsa + ggtitle("Aligned on regions left boundary. CNE is 50/48. Okinawa - Osaka")
Okinawa–Barcelona
pOkiBar <- twoBySixFigure(
computePlotData_simplified("Oki", "Bar", reps = reps, CE = ces$ce__50__48, direction = "right"),
computePlotData_simplified("Oki", "Bar", annots = annots, operons = operons, direction = "right", withGenes = "only"),
computePlotData_simplified("Oki", "Bar", annots = annots, direction = "right", withGenes = "no")
) +
ggtitle("Okinawa - Barcelona")Okinawa–Kume
pOkiKum <- twoBySixFigure(
computePlotData_simplified("Oki", "Kum", reps = reps, CE = ces$ce__50__48, direction = "right"),
computePlotData_simplified("Oki", "Kum", annots = annots, operons = operons, direction = "right", withGenes = "only"),
computePlotData_simplified("Oki", "Kum", annots = annots, direction = "right", withGenes = "no")
) +
ggtitle("Okinawa - Kum")## Warning in valid.GenomicRanges.seqinfo(x, suggest.trim = TRUE): GRanges object contains 1 out-of-bound range located on sequence PAR.
## Note that ranges located on a sequence whose length is unknown (NA) or
## on a circular sequence are not considered out-of-bound (use
## seqlengths() and isCircular() to get the lengths and circularity flags
## of the underlying sequences). You can use trim() to trim these ranges.
## See ?`trim,GenomicRanges-method` for more information.
## Warning in valid.GenomicRanges.seqinfo(x, suggest.trim = TRUE): GRanges object contains 1 out-of-bound range located on sequence PAR.
## Note that ranges located on a sequence whose length is unknown (NA) or
## on a circular sequence are not considered out-of-bound (use
## seqlengths() and isCircular() to get the lengths and circularity flags
## of the underlying sequences). You can use trim() to trim these ranges.
## See ?`trim,GenomicRanges-method` for more information.
## Warning in valid.GenomicRanges.seqinfo(x, suggest.trim = TRUE): GRanges object contains 1 out-of-bound range located on sequence PAR.
## Note that ranges located on a sequence whose length is unknown (NA) or
## on a circular sequence are not considered out-of-bound (use
## seqlengths() and isCircular() to get the lengths and circularity flags
## of the underlying sequences). You can use trim() to trim these ranges.
## See ?`trim,GenomicRanges-method` for more information.
Osaka–Okinawa
pOsaOki <- twoBySixFigure(
computePlotData_simplified("Osa", "Oki", reps = reps, CE = ces$ce__50__48, direction = "right"),
computePlotData_simplified("Osa", "Oki", annots = annots, operons = operons, direction = "right", withGenes = "only"),
computePlotData_simplified("Osa", "Oki", annots = annots, direction = "right", withGenes = "no")
) +
ggtitle("Osaka - Okinawa")Osaka–Barcelona
pOsaBar <- twoBySixFigure(
computePlotData_simplified("Osa", "Bar", reps = reps, CE = ces$ce__50__48, direction = "right"),
computePlotData_simplified("Osa", "Bar", annots = annots, operons = operons, direction = "right", withGenes = "only"),
computePlotData_simplified("Osa", "Bar", annots = annots, direction = "right", withGenes = "no")
) +
ggtitle("Osaka - Barcelona")## Warning in valid.GenomicRanges.seqinfo(x, suggest.trim = TRUE): GRanges object contains 1 out-of-bound range located on sequence Chr1.
## Note that ranges located on a sequence whose length is unknown (NA) or
## on a circular sequence are not considered out-of-bound (use
## seqlengths() and isCircular() to get the lengths and circularity flags
## of the underlying sequences). You can use trim() to trim these ranges.
## See ?`trim,GenomicRanges-method` for more information.
## Warning in valid.GenomicRanges.seqinfo(x, suggest.trim = TRUE): GRanges object contains 1 out-of-bound range located on sequence Chr1.
## Note that ranges located on a sequence whose length is unknown (NA) or
## on a circular sequence are not considered out-of-bound (use
## seqlengths() and isCircular() to get the lengths and circularity flags
## of the underlying sequences). You can use trim() to trim these ranges.
## See ?`trim,GenomicRanges-method` for more information.
## Warning in valid.GenomicRanges.seqinfo(x, suggest.trim = TRUE): GRanges object contains 1 out-of-bound range located on sequence Chr1.
## Note that ranges located on a sequence whose length is unknown (NA) or
## on a circular sequence are not considered out-of-bound (use
## seqlengths() and isCircular() to get the lengths and circularity flags
## of the underlying sequences). You can use trim() to trim these ranges.
## See ?`trim,GenomicRanges-method` for more information.Osaka–Aomori
pOsaAom <- twoBySixFigure(
computePlotData_simplified("Osa", "Aom", reps = reps, CE = ces$ce__50__48, direction = "right"),
computePlotData_simplified("Osa", "Aom", annots = annots, operons = operons, direction = "right", withGenes = "only"),
computePlotData_simplified("Osa", "Aom", annots = annots, direction = "right", withGenes = "no")
) +
ggtitle("Osaka - Aomori")## Warning in valid.GenomicRanges.seqinfo(x, suggest.trim = TRUE): GRanges object contains 2 out-of-bound ranges located on sequences Chr1
## and Chr2. Note that ranges located on a sequence whose length is
## unknown (NA) or on a circular sequence are not considered out-of-bound
## (use seqlengths() and isCircular() to get the lengths and circularity
## flags of the underlying sequences). You can use trim() to trim these
## ranges. See ?`trim,GenomicRanges-method` for more information.
## Warning in valid.GenomicRanges.seqinfo(x, suggest.trim = TRUE): GRanges object contains 2 out-of-bound ranges located on sequences Chr1
## and Chr2. Note that ranges located on a sequence whose length is
## unknown (NA) or on a circular sequence are not considered out-of-bound
## (use seqlengths() and isCircular() to get the lengths and circularity
## flags of the underlying sequences). You can use trim() to trim these
## ranges. See ?`trim,GenomicRanges-method` for more information.
## Warning in valid.GenomicRanges.seqinfo(x, suggest.trim = TRUE): GRanges object contains 2 out-of-bound ranges located on sequences Chr1
## and Chr2. Note that ranges located on a sequence whose length is
## unknown (NA) or on a circular sequence are not considered out-of-bound
## (use seqlengths() and isCircular() to get the lengths and circularity
## flags of the underlying sequences). You can use trim() to trim these
## ranges. See ?`trim,GenomicRanges-method` for more information.
Extra genomic feature coverage plots.
Bridge regions classified by width.
Short bridge regions contain small introns. Long bridge regions contain repeats, which may be in introns.
computePlotData_for_bridge <- function(target, query, annots=NULL, reps=NULL, CE=NULL, tfbs=NULL, operons=NULL, direction="left") {
pair <- paste(target, query, sep = "_")
current_GB_bri <- bri[[pair]]
current_GB_bri_small <- current_GB_bri[width(current_GB_bri) < 150]
current_GB_bri_long <- current_GB_bri[width(current_GB_bri) >= 150]
targetGenome <- unique(genome(gbs[[pair]]))
prepareFreqPlot_default <- \(x) prepareFreqPlot(x, annot = annots[[target]], rep = reps[[targetGenome]], cne = CE[[pair]], tfbs = tfbs, operons = operons[[target]], direction = direction)
hmList <- SimpleList()
hmList$bri <- prepareFreqPlot_default(current_GB_bri)
hmList$bri_small <- prepareFreqPlot_default(current_GB_bri_small)
hmList$bri_long <- prepareFreqPlot_default(current_GB_bri_long)
hmList
}
hmList_to_plot_noncoding <- computePlotData_for_bridge("Oki", "Osa", reps = reps, CE = ces$ce__50__48)
hmList_to_plot_coding <- computePlotData_for_bridge("Oki", "Osa", annots = annots, operons = operons)
list_of_heatmaps_to_long_tibble <- function(hmList_to_plot, desc = NULL) {
tib <-rbind(
hmList_to_plot$bri |> prepareGGFreqPlot("01_bridge"),
hmList_to_plot$bri_small |> prepareGGFreqPlot("02_bridge_small"),
hmList_to_plot$bri_long |> prepareGGFreqPlot("02_bridge_long")
)
tib$desc2 <- desc
tib
}
rbind(
list_of_heatmaps_to_long_tibble(hmList_to_plot_noncoding, "noncoding"),
list_of_heatmaps_to_long_tibble(hmList_to_plot_coding, "coding")
) |> ggplot() +
aes(pos, freq, col = what) +
geom_line() +
facet_wrap(~desc2 + desc, nrow = 2) +
theme_bw() +
ggtitle("Aligned on regions left boundary. CNE is 50/48. Okinawa - Osaka")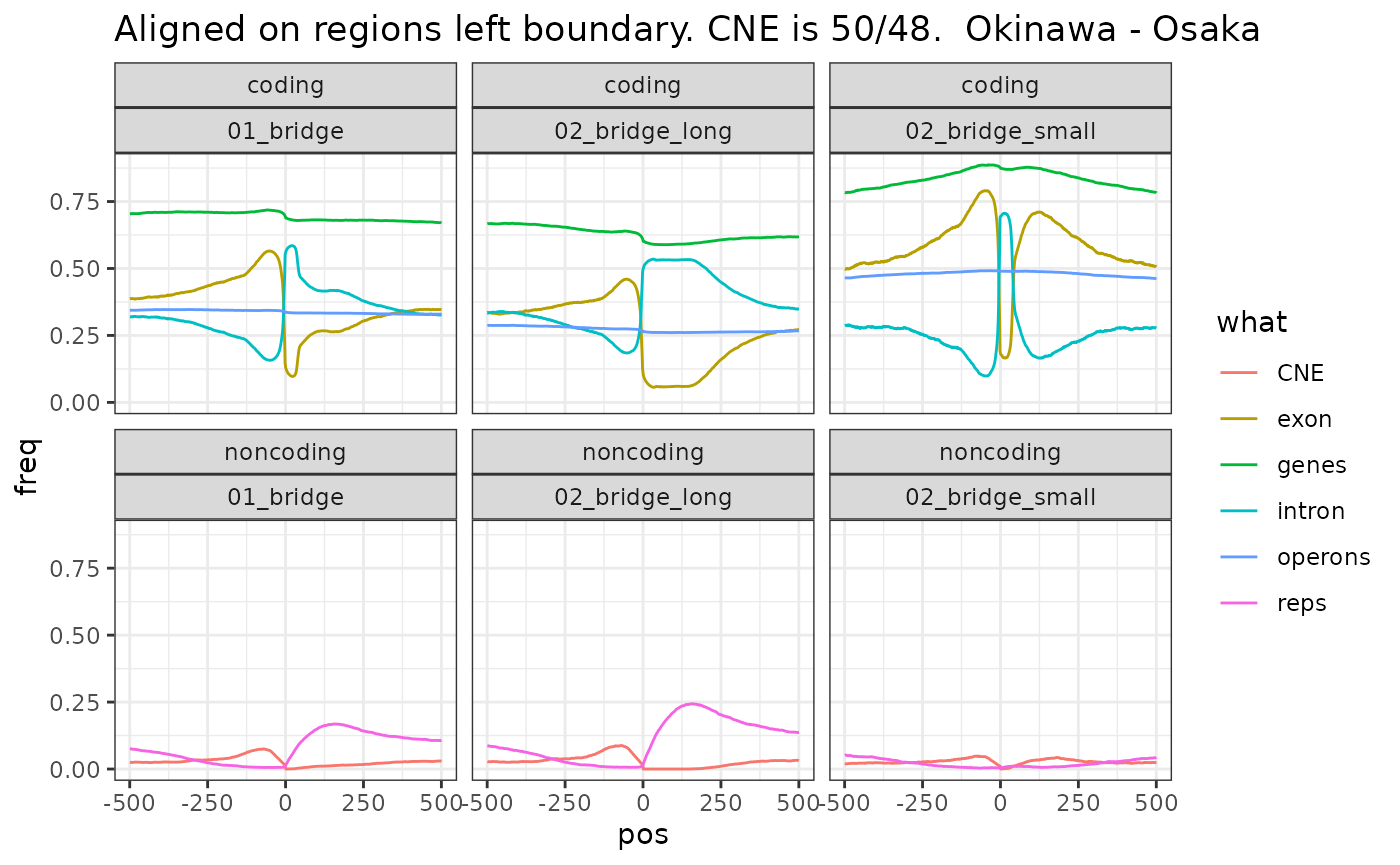
Aligning on center instead of border.
hmList_to_plot.centered <- computePlotData("Oki", "Osa", annots = annots, CE = ces$ce__50__48, tfbs = tfbs, operons = operons, direction = "mid")
(p_boundary <- rbind(
hmList_to_plot.centered$aln_nonCoa |> prepareGGFreqPlot("01_isolated_alns"),
hmList_to_plot.centered$unmap |> prepareGGFreqPlot("02_unmapped"),
hmList_to_plot.centered$syn_block |> prepareGGFreqPlot("03_colinear_reg"),
hmList_to_plot.centered$bri |> prepareGGFreqPlot("04_bridge"),
hmList_to_plot.centered$aln_coa |> prepareGGFreqPlot("05_colinear_alns")
)|> ggplot() +
aes(pos, freq, col = what) +
geom_line() +
facet_wrap(~desc, nrow = 1) +
theme_bw() +
ggtitle("Aligned on center of regions. CNE is 50/48. Okinawa - Osaka"))
Splice junctions
rbind(
feature_coverage(gbs$Oki_Osa[gbs$Oki_Osa$nonCoa], intronicParts(annots$Oki) |> resize(1), window = 1000, lab = "splicejunctions", dir = "left") |>
Heatmap_to_freq_tibble("01_isolated"),
feature_coverage(unal$Oki_Osa[unal$Oki_Osa$nonCoa], intronicParts(annots$Oki) |> resize(1), window = 1000, lab = "splicejunctions", dir = "left") |>
Heatmap_to_freq_tibble("02_unaligned"),
feature_coverage(coa$Oki_Osa[!coa$Oki_Osa$nonCoa], intronicParts(annots$Oki) |> resize(1), window = 1000, lab = "splicejunctions", dir = "left") |>
Heatmap_to_freq_tibble("03_colinear_reg"),
feature_coverage(bri$Oki_Osa, intronicParts(annots$Oki) |> resize(1), window = 1000, lab = "splicejunctions", dir = "left") |>
Heatmap_to_freq_tibble("04_bridge")
) |> ggplot() + aes(pos, freq, col = desc) + geom_line()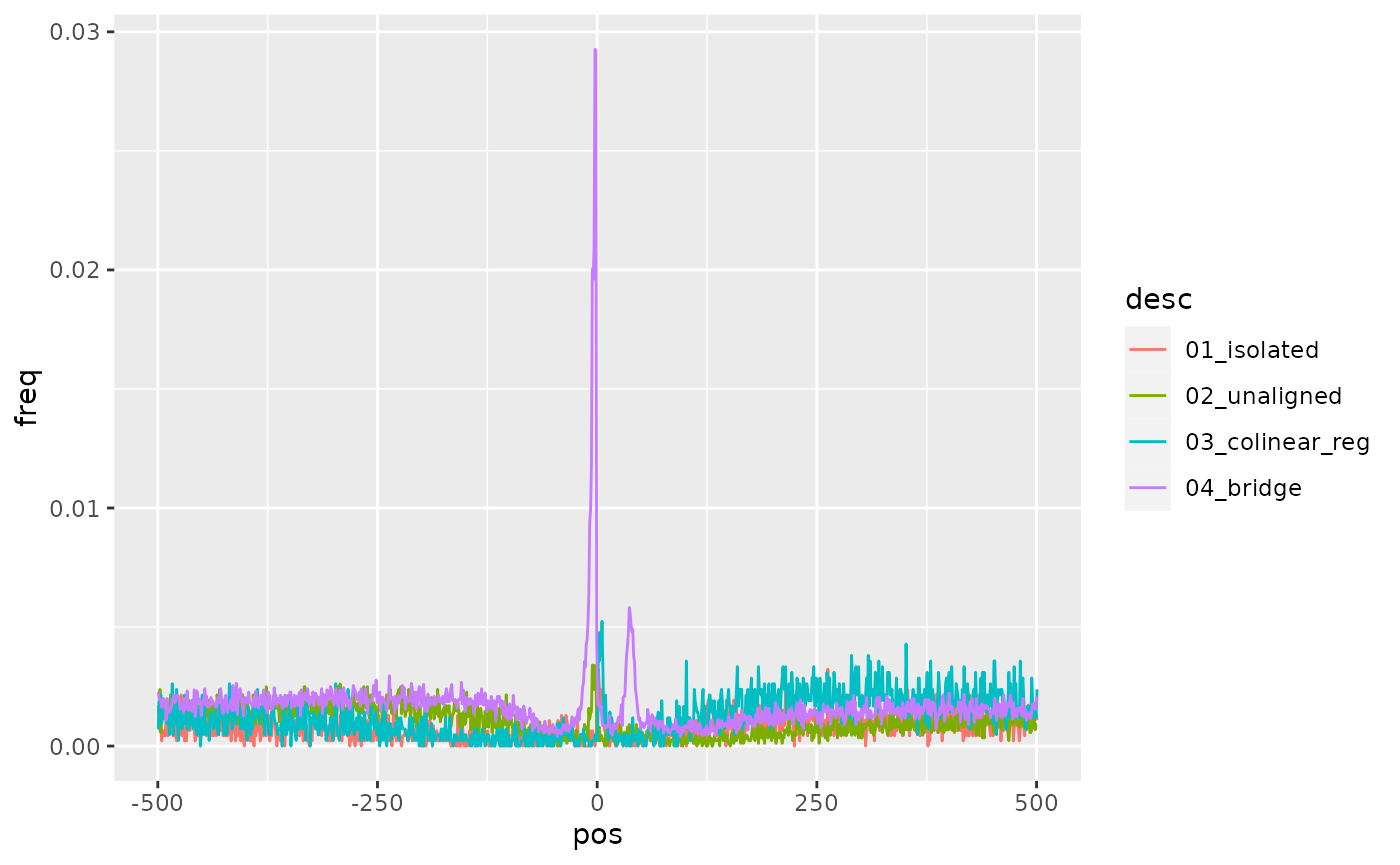
PWM
The PWM motif, which is rare in comparison to the other features, follows a profile that bears resemblance to the repeat profile.
gadems.invLeftGap <- readRDS("gadems.invLeftGap.Rda")
pwm <- gadems.invLeftGap$Oki_Osa[[1]]
pwm@consensus## [1] "rArAAGCCGCdwAGCsGCw"
# pwmHits <- matchPWM(pwm = sapply(pwm@alignList, \(x) x@seq) |> as("DNAStringSet") |> PWM(), genomes$Oki) # Gives different scores...
pwmHits <- matchPWM(pwm = rGADEM::getPWM(pwm), genomes$OKI2018.I69) |> suppressWarnings()
# Suppressing warnings like:
# Warning messages:
# 1: In .Call2("XString_match_PWM", pwm, subject, min.score, count.only, :
# 'subject' contains letters not in [ACGT] ==> assigned weight 0 to them
#
# plotHeatmapMeta(list(feature_coverage(current_GB_wide, pwmHits, win = 1e3, lab = "wide", direction = "left")))
# plotHeatmapMeta(list(feature_coverage(current_GB_narrow, pwmHits, win = 1e3, lab = "narrow", direction = "left")))Gene feature coverage around PWM hits
Let’s look at coverage around PWM hits of the AAGCsGCwwmkCGrCTTyn motif
current_GB <- pwmHits
hmList_PWM_OKI <- list()
hmList_PWM_OKI$genes <- feature_coverage(current_GB, genes(annots$Oki), win = 2000, lab = "Oki genes", direction = "left")
hmList_PWM_OKI$promoters <- feature_coverage(current_GB, promoters(annots$Oki), win = 2000, lab = "Oki promoters", direction = "left")## Warning in valid.GenomicRanges.seqinfo(x, suggest.trim = TRUE): GRanges object contains 4 out-of-bound ranges located on sequences
## chr1, chrUn_1, and chrUn_10. Note that ranges located on a sequence
## whose length is unknown (NA) or on a circular sequence are not
## considered out-of-bound (use seqlengths() and isCircular() to get the
## lengths and circularity flags of the underlying sequences). You can use
## trim() to trim these ranges. See ?`trim,GenomicRanges-method` for more
## information.
hmList_PWM_OKI$exons <- feature_coverage(current_GB, exonicParts(annots$Oki), win = 2000, lab = "Oki exons", direction = "left")
hmList_PWM_OKI$introns <- feature_coverage(current_GB, intronicParts(annots$Oki), win = 2000, lab = "Oki introns", direction = "left")
hmList_PWM_OKI$cds <- feature_coverage(current_GB, cds(annots$Oki), win = 2000, lab = "Oki cds", direction = "left")
plotHeatmapMeta(hmList_PWM_OKI[c("genes", "exons", "cds")]) # Show they look alike. We keep "exons"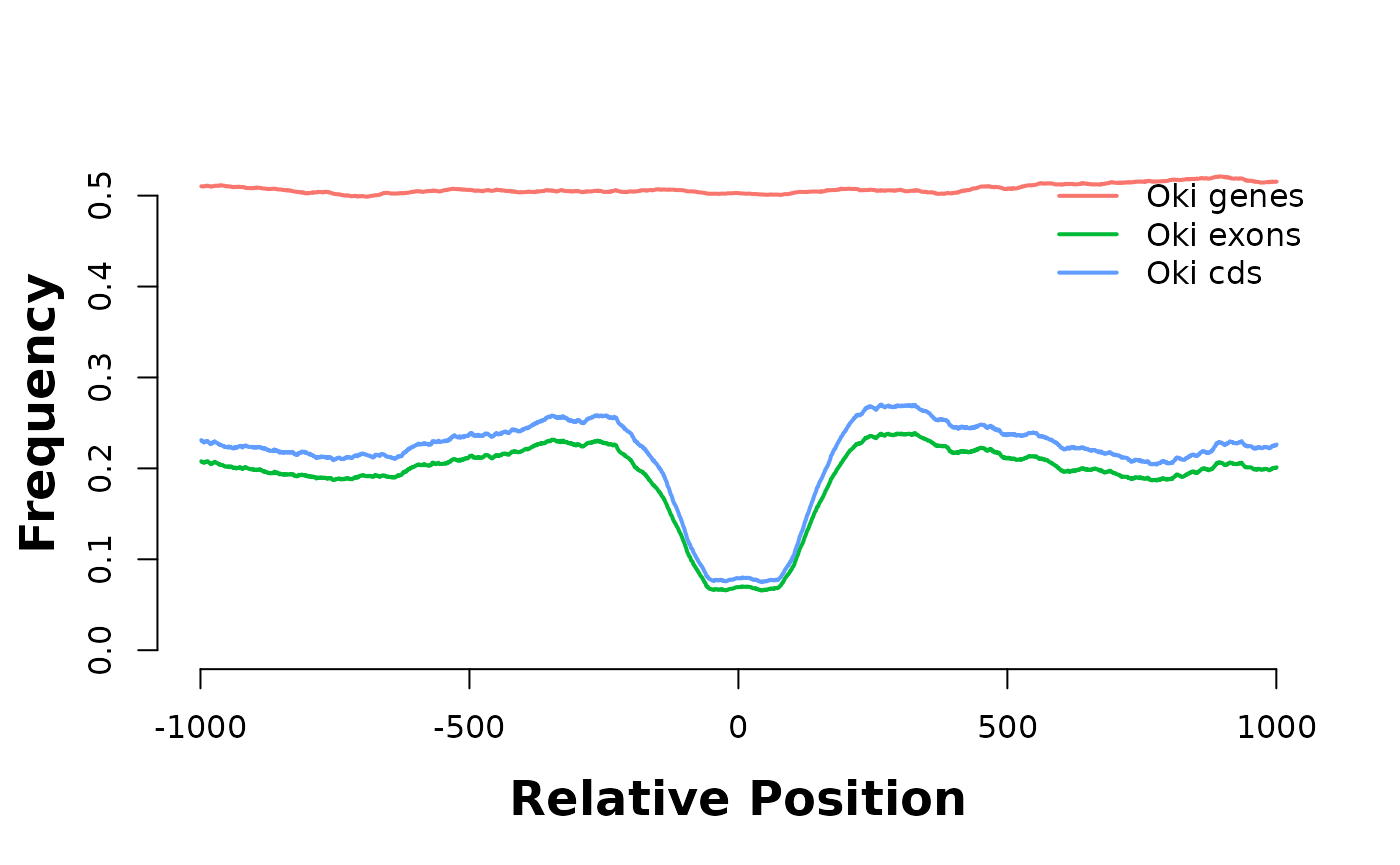
plotHeatmapMeta(hmList_PWM_OKI[c("promoters", "exons", "introns")])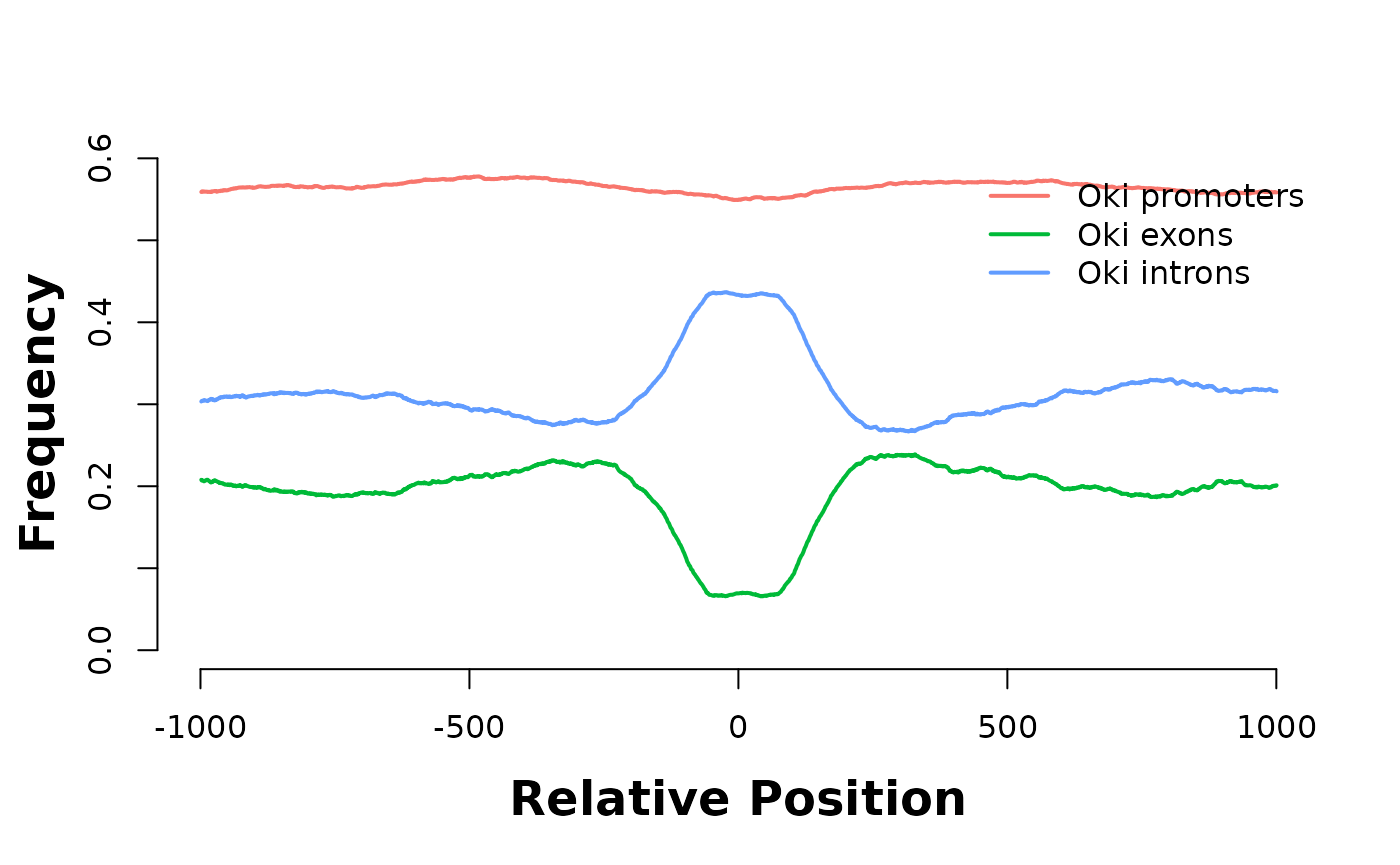
They are enriched in introns…
#
# # Sandbox to make sanity checks.
#
# a <- coa$Oki_Osa
# b <- unlist(fiveUTRsByTranscript(annots$Oki))
#
# hm_a <- feature_coverage(a, b, win = 2000, lab = "All ranges", direction = "left")
# hm_b <- feature_coverage(subsetByOverlaps(a, b), b, win = 2000, lab = "Subsetted ranges", direction = "left")
#
# plotHeatmapMeta(list(hm_a))
# plotHeatmapMeta(list(hm_b))Breakpoint coverage around PWM hits
TODO: triplecheck feature_coverage, it is old…
current_GB <- pwmHits
hmList_PWM_OKI$gbOsa <- feature_coverage(current_GB, gbs$Oki_Osa, win = 5000, lab = "Oki-Osa", direction = "left")
hmList_PWM_OKI$gbBar <- feature_coverage(current_GB, gbs$Oki_Bar, win = 5000, lab = "Oki-Bar", direction = "left")
hmList_PWM_OKI$gbKum <- feature_coverage(current_GB, gbs$Oki_Kum, win = 5000, lab = "Oki-Kum", direction = "left")
plotHeatmapMeta(hmList_PWM_OKI[c("gbOsa", "gbBar", "gbKum")])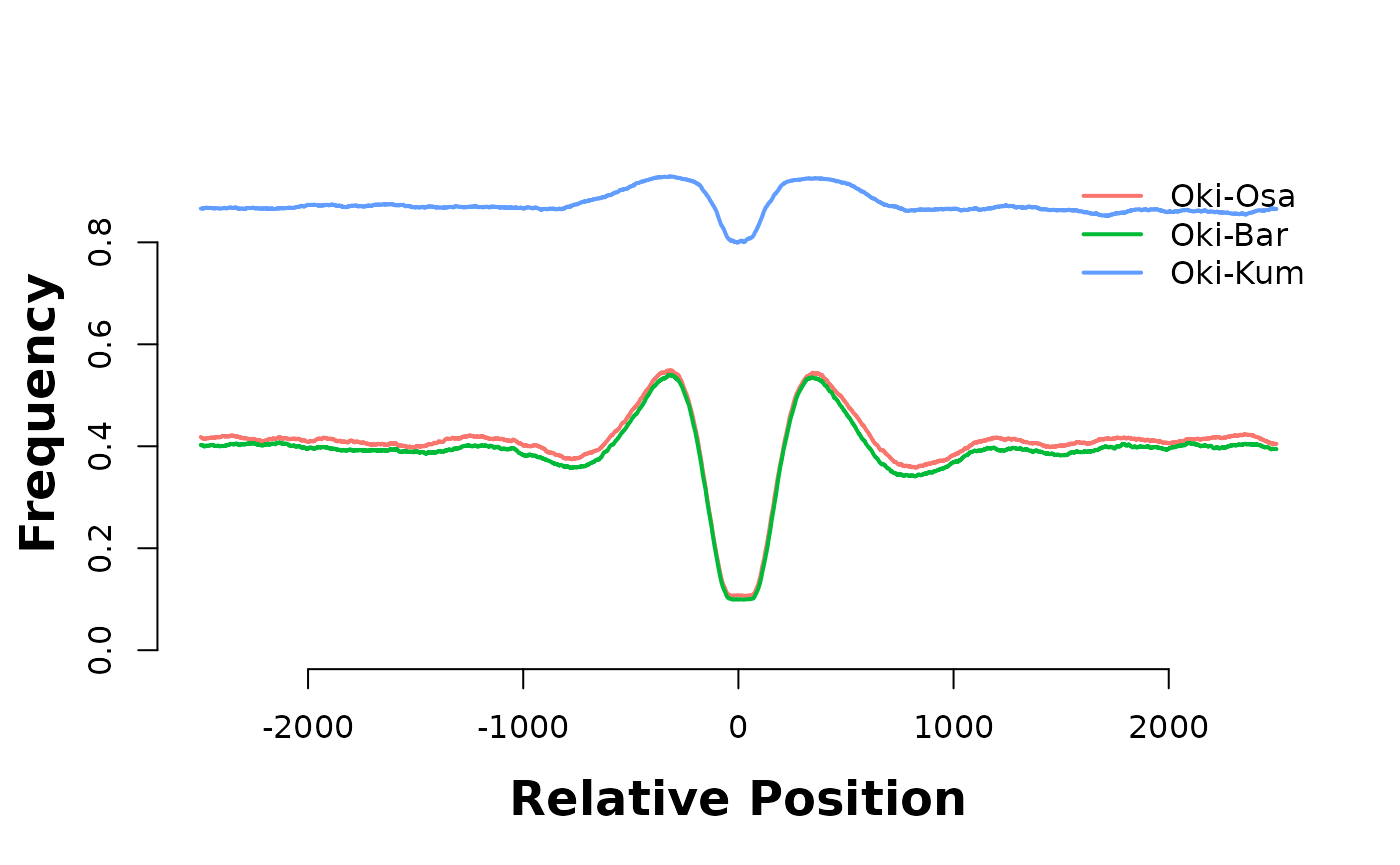
hmList_PWM_OKI$coOsa <- feature_coverage(current_GB, coa$Oki_Osa, win = 5000, lab = "Oki-Osa", direction = "left")
hmList_PWM_OKI$coBar <- feature_coverage(current_GB, coa$Oki_Bar, win = 5000, lab = "Oki-Bar", direction = "left")
hmList_PWM_OKI$coKum <- feature_coverage(current_GB, coa$Oki_Kum, win = 5000, lab = "Oki-Kum", direction = "left")
plotHeatmapMeta(hmList_PWM_OKI[c("coOsa", "coBar", "coKum")])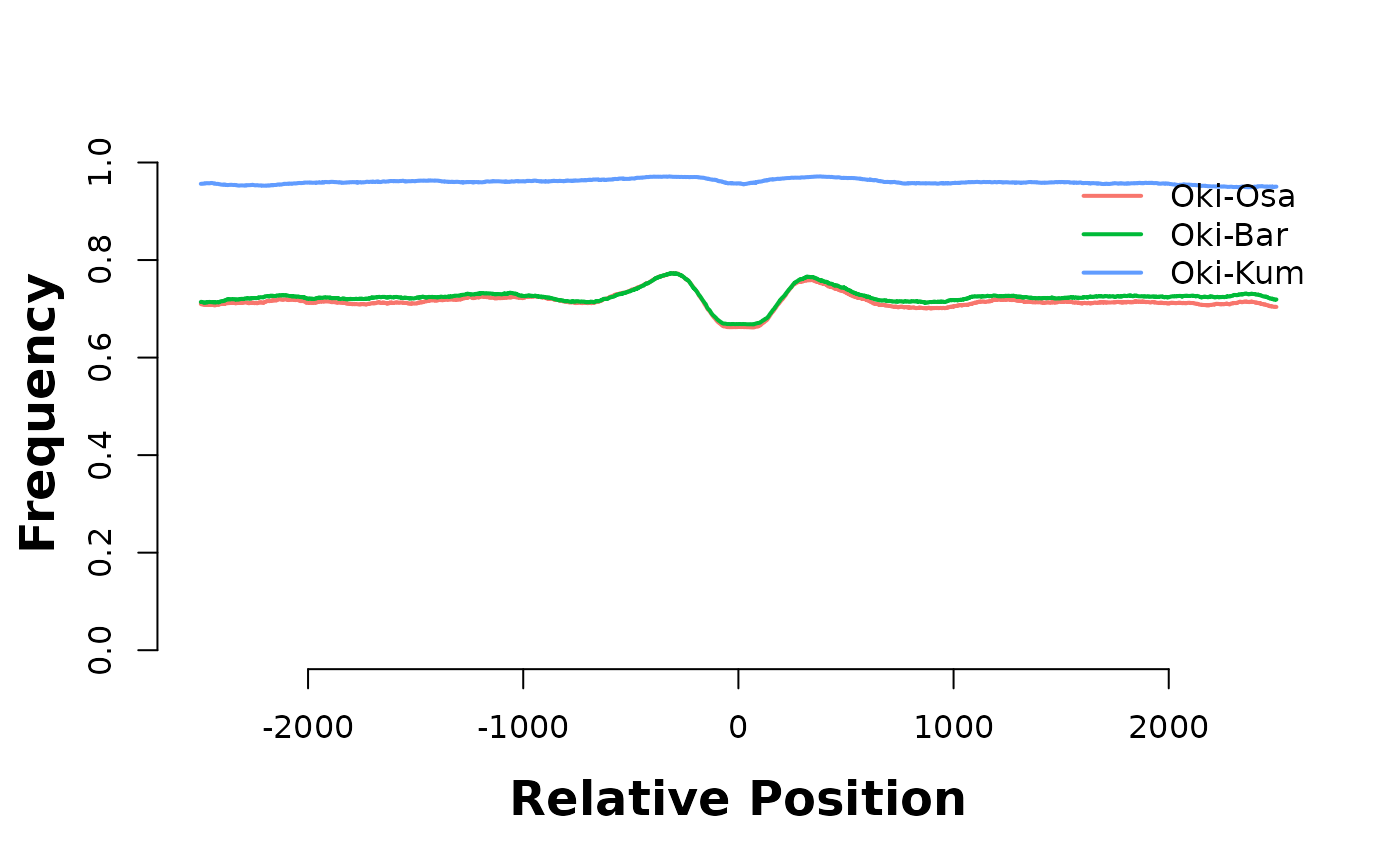
Okinawa genome coverage
As previously discussed, low coverage over an alignment stop could
lower the likelihood of it being considered as a breakpoint. We have
per-base coverage depth information for the Okinawan genome
(Oki_cov_pb). However, the coverage of this particular
assembly is quite good. In fact, we can investigate the coverge over
alignment stops from the information obtained using
master_bp_analysis.
# fin_Oki <- fin_gr_O_Oki[[2]]
# min_cov <- min(min(fin_Oki$left_cov_pb), min(fin_Oki$right_cov_pb)) # minimum coverage over an alignment stop
# min_cov
# length(fin_Oki[fin_Oki$left_cov_pb == min_cov]) + length(fin_Oki[fin_Oki$right_cov_pb == min_cov]) # how many of the minimum coverage is observed
# length(fin_Oki[fin_Oki$left_cov_pb <= 50]) + length(fin_Oki[fin_Oki$right_cov_pb <= 50]) # how many alignment stops have a coverage of less than or euqal to 50We may choose to kick out the one alignment stop for which there is no coverage. However, only 15 out of 34572 alignment stops have a coverage of less than or equal to 50, meaning that it would be hard to exclude more than just a few alignment stops using coverage information for breakpoint analysis.
Operons and genes
Do alignments often cross gene or operon boundaries ?
Operons boundaries
Operon boundaries tend to coincide with alignment boundaries and the largest shift in frequency is for colinear alignments. Baseline frequency of colinear regions is higher than 0.6 on both side of operon boundaries.
feature_coverage_on_operons <- {
function(feat, desc)
feature_coverage(operons$Oki, feat, window = 1000, lab = "", dir = "left") |> Heatmap_to_freq_tibble(desc)
}
pairname <- "Oki_Bar"
pairname <- "Oki_Osa"
p_op_scrambling <- rbind(
feature_coverage_on_operons(gbs[[pairname]][gbs[[pairname]]$nonCoa], "01_isolated"),
feature_coverage_on_operons(unal[[pairname]][unal[[pairname]]$nonCoa], "02_unaligned"),
feature_coverage_on_operons(gbs[[pairname]][!gbs[[pairname]]$nonCoa], "03_colinear_aln"),
#feature_coverage_on_operons(subsetByOverlaps(gbs$Oki_Osa[!gbs$Oki_Osa$nonCoa], granges(coa$Oki_Osa[!coa$Oki_Osa$nonCoa] - 1),type = "within"), "03b_colinear_aln_intern"),
#feature_coverage_on_operons(subsetByOverlaps(gbs$Oki_Osa[!gbs$Oki_Osa$nonCoa], unal$Oki_Osa[unal$Oki_Osa$nonCoa] + 1), "03c_colinear_aln_extern"),
feature_coverage_on_operons(bri[[pairname]], "04_bridge"),
feature_coverage_on_operons(coa[[pairname]][!coa[[pairname]]$nonCoa], "05_colinear_reg")
) |> ggplot() + aes(pos, freq, col = desc) + geom_line() + ggtitle("operons", subtitle = "n = 2017") + theme_bw()Gene boundaries inside operons
# Shrinking operons of 1 base and requesting full overlap
# in order to exclude the genes at the boundaries.
genes_inside_operons <- subsetByOverlaps(genes(annots$Oki), operons$Oki -1, type = "within")
genes_outside_operons <- subsetByOverlaps(genes(annots$Oki), cleanGaps(operons$Oki), type = "within")
feature_coverage_on_genes <- {
function(feat, desc)
feature_coverage(genes_inside_operons, feat, window = 1000, lab = "", dir = "left") |> Heatmap_to_freq_tibble(desc)
}
p_gene_int_scrambling <- rbind(
feature_coverage_on_genes(gbs$Oki_Osa[gbs$Oki_Osa$nonCoa], "01_isolated"),
feature_coverage_on_genes(unal$Oki_Osa[unal$Oki_Osa$nonCoa], "02_unmapped"),
feature_coverage_on_genes(gbs$Oki_Osa[!gbs$Oki_Osa$nonCoa], "03_colinear_aln"),
feature_coverage_on_genes(bri$Oki_Osa, "04_bridge"),
feature_coverage_on_genes(coa$Oki_Osa[!coa$Oki_Osa$nonCoa], "05_colinear_reg")
) |> ggplot() + aes(pos, freq, col = desc) + geom_line() + ggtitle("genes_internal_to_operons", subtitle = "n = 1348") + theme_bw()
feature_coverage_on_genes_outside <- {
function(feat, desc)
feature_coverage(genes_outside_operons, feat, window = 1000, lab = "", dir = "left") |> Heatmap_to_freq_tibble(desc)
}
p_gene_ext_scrambling <- rbind(
feature_coverage_on_genes_outside(gbs$Oki_Osa[gbs$Oki_Osa$nonCoa], "01_isolated"),
feature_coverage_on_genes_outside(unal$Oki_Osa[unal$Oki_Osa$nonCoa], "02_unmapped"),
feature_coverage_on_genes_outside(gbs$Oki_Osa[!gbs$Oki_Osa$nonCoa], "03_colinear_aln"),
feature_coverage_on_genes_outside(bri$Oki_Osa, "04_bridge"),
feature_coverage_on_genes_outside(coa$Oki_Osa[!coa$Oki_Osa$nonCoa], "05_colinear_reg")
) |> ggplot() + aes(pos, freq, col = desc) + geom_line() + ggtitle("genes_outside_operons", subtitle = "n = 11688") + theme_bw()Summary plot
p_gene_ext_scrambling + p_op_scrambling + p_gene_int_scrambling + plot_layout(nrow = 1, guides = 'collect')
Operon conservation
# Distribution of operon lengths
operons$Oki$n |> table()##
## 2 3 4 5 6 7 8 9 10 11 14
## 1943 647 259 134 69 33 19 10 6 3 1
# An operon is scrambled if it overlaps with an unaligned noncoalesced (= unmapped) region
scrambled_operons <- subsetByOverlaps(operons$Oki, unal$Oki_Osa[unal$Oki_Osa$nonCoa])
not_scrambled_operons <- operons$Oki[! operons$Oki %in% scrambled_operons ]
# Distribution of operon lengths (scrambled)
scrambled_operons$n |> table()##
## 2 3 4 5 6 7 8 9 10 11
## 905 298 106 44 31 9 7 6 4 3
not_scrambled_operons$n |> table()##
## 2 3 4 5 6 7 8 9 10 14
## 1038 349 153 90 38 24 12 4 2 1
# Example of long scrambled operons
scrambled_operons[scrambled_operons$n > 5]## GRanges object with 60 ranges and 2 metadata columns:
## seqnames ranges strand | n gene_id
## <Rle> <IRanges> <Rle> | <integer> <CharacterList>
## [1] chr1 5715727-5730794 + | 7 g1521,g1522,g1523,...
## [2] chr1 6147428-6157837 + | 6 g1613,g1614,g1615,...
## [3] chr1 9809914-9836205 + | 10 g2620,g2621,g2622,...
## [4] chr1 10022216-10035952 + | 7 g2668,g2669,g2670,...
## [5] chr1 10547772-10569235 - | 9 g2832,g2833,g2834,...
## ... ... ... ... . ... ...
## [56] XSR 6611822-6622862 + | 7 g15207,g15208,g15209,...
## [57] XSR 6843015-6861410 + | 8 g15282,g15283,g15284,...
## [58] XSR 7875989-7897781 - | 7 g15606,g15607,g15608,...
## [59] XSR 10693937-10705412 + | 6 g16499,g16500,g16501,...
## [60] XSR 12736159-12757464 - | 8 g17016,g17017,g17018,...
## -------
## seqinfo: 19 sequences from OKI2018.I69 genomeIn comparison: genes
all_genes <- genes(annots$Oki)
scrambled_genes <- (subsetByOverlaps(all_genes, unal$Oki_Osa[unal$Oki_Osa$nonCoa])) |> sort(i=F) |> flagLongShort(longShort$OKI2018.I69)
not_scrambled_genes <- all_genes[! all_genes %in% scrambled_genes] |> sort(i=F) |> flagLongShort(longShort$OKI2018.I69)
length(scrambled_genes)## [1] 5294
length(all_genes)## [1] 17291
scr_g_tbl <- table(paste(seqnames( scrambled_genes), scrambled_genes$Arm))
nsc_g_tbl <- table(paste(seqnames(not_scrambled_genes), not_scrambled_genes$Arm))
Oki_Osa_scr_ratio_long_arm <- c(
scr_g_tbl[["chr1 long"]] / ( scr_g_tbl[["chr1 long"]] + nsc_g_tbl[["chr1 long"]]),
scr_g_tbl[["chr2 long"]] / ( scr_g_tbl[["chr2 long"]] + nsc_g_tbl[["chr2 long"]]),
scr_g_tbl[["PAR long"]] / ( scr_g_tbl[["PAR long"]] + nsc_g_tbl[["PAR long"]])
)
mean(Oki_Osa_scr_ratio_long_arm)## [1] 0.2314521
Oki_Osa_scr_ratio_short_arm <- c(
scr_g_tbl[["chr1 short"]] / ( scr_g_tbl[["chr1 short"]] + nsc_g_tbl[["chr1 short"]]),
scr_g_tbl[["chr2 short"]] / ( scr_g_tbl[["chr2 short"]] + nsc_g_tbl[["chr2 short"]]),
scr_g_tbl[["PAR short"]] / ( scr_g_tbl[["PAR short"]] + nsc_g_tbl[["PAR short"]])
)
mean(Oki_Osa_scr_ratio_short_arm)## [1] 0.5374672
t.test(Oki_Osa_scr_ratio_long_arm, Oki_Osa_scr_ratio_short_arm, paired = T)##
## Paired t-test
##
## data: Oki_Osa_scr_ratio_long_arm and Oki_Osa_scr_ratio_short_arm
## t = -9.5775, df = 2, p-value = 0.01073
## alternative hypothesis: true mean difference is not equal to 0
## 95 percent confidence interval:
## -0.4434904 -0.1685397
## sample estimates:
## mean difference
## -0.3060151Replicate on an independent pair of genomes.
all_genes <- genes(annots$Bar)
scrambled_genes <- (subsetByOverlaps(all_genes, unal$Bar_Kum[unal$Bar_Kum$nonCoa])) |> sort(i=F) |> flagLongShort(longShort$Bar2.p4)
not_scrambled_genes <- all_genes[! all_genes %in% scrambled_genes] |> sort(i=F) |> flagLongShort(longShort$Bar2.p4)
length(scrambled_genes)## [1] 4002
length(all_genes)## [1] 14272
scr_g_tbl <- table(paste(seqnames( scrambled_genes), scrambled_genes$Arm))
nsc_g_tbl <- table(paste(seqnames(not_scrambled_genes), not_scrambled_genes$Arm))
Bar_Kum_scr_ratio_long_arm <- c(
scr_g_tbl[["Chr1 long"]] / ( scr_g_tbl[["Chr1 long"]] + nsc_g_tbl[["Chr1 long"]]),
scr_g_tbl[["Chr2 long"]] / ( scr_g_tbl[["Chr2 long"]] + nsc_g_tbl[["Chr2 long"]]),
scr_g_tbl[["PAR long"]] / ( scr_g_tbl[["PAR long"]] + nsc_g_tbl[["PAR long"]])
)
Bar_Kum_scr_ratio_short_arm <- c(
scr_g_tbl[["Chr1 short"]] / ( scr_g_tbl[["Chr1 short"]] + nsc_g_tbl[["Chr1 short"]]),
scr_g_tbl[["Chr2 short"]] / ( scr_g_tbl[["Chr2 short"]] + nsc_g_tbl[["Chr2 short"]]),
scr_g_tbl[["PAR short"]] / ( scr_g_tbl[["PAR short"]] + nsc_g_tbl[["PAR short"]])
)
t.test(Bar_Kum_scr_ratio_long_arm, Bar_Kum_scr_ratio_short_arm, paired = TRUE)##
## Paired t-test
##
## data: Bar_Kum_scr_ratio_long_arm and Bar_Kum_scr_ratio_short_arm
## t = -16.994, df = 2, p-value = 0.003445
## alternative hypothesis: true mean difference is not equal to 0
## 95 percent confidence interval:
## -0.3825454 -0.2279699
## sample estimates:
## mean difference
## -0.3052576## [1] 0.2055334## [1] 0.5111697
t.test(
c(Oki_Osa_scr_ratio_long_arm, Bar_Kum_scr_ratio_long_arm),
c(Oki_Osa_scr_ratio_short_arm, Bar_Kum_scr_ratio_short_arm),
paired = TRUE
)##
## Paired t-test
##
## data: c(Oki_Osa_scr_ratio_long_arm, Bar_Kum_scr_ratio_long_arm) and c(Oki_Osa_scr_ratio_short_arm, Bar_Kum_scr_ratio_short_arm)
## t = -18.644, df = 5, p-value = 8.172e-06
## alternative hypothesis: true mean difference is not equal to 0
## 95 percent confidence interval:
## -0.3477765 -0.2634962
## sample estimates:
## mean difference
## -0.3056363How about exons ?
all_exons <- exonicParts(annots$Oki)
scrambled_exons <- (subsetByOverlaps(all_exons, unal$Oki_Osa[unal$Oki_Osa$nonCoa])) |> sort(i=F) |> flagLongShort(longShort$OKI2018.I69)
not_scrambled_exons <- all_exons[! all_exons %in% scrambled_exons] |> sort(i=F) |> flagLongShort(longShort$OKI2018.I69)
length(scrambled_exons)## [1] 16787
length(all_exons)## [1] 106811
scr_g_tbl <- table(paste(seqnames( scrambled_exons), scrambled_exons$Arm))
nsc_g_tbl <- table(paste(seqnames(not_scrambled_exons), not_scrambled_exons$Arm))
Oki_Osa_scr_ratio_long_arm <- c(
scr_g_tbl[["chr1 long"]] / ( scr_g_tbl[["chr1 long"]] + nsc_g_tbl[["chr1 long"]]),
scr_g_tbl[["chr2 long"]] / ( scr_g_tbl[["chr2 long"]] + nsc_g_tbl[["chr2 long"]]),
scr_g_tbl[["PAR long"]] / ( scr_g_tbl[["PAR long"]] + nsc_g_tbl[["PAR long"]])
)
mean(Oki_Osa_scr_ratio_long_arm)## [1] 0.1172023
Oki_Osa_scr_ratio_short_arm <- c(
scr_g_tbl[["chr1 short"]] / ( scr_g_tbl[["chr1 short"]] + nsc_g_tbl[["chr1 short"]]),
scr_g_tbl[["chr2 short"]] / ( scr_g_tbl[["chr2 short"]] + nsc_g_tbl[["chr2 short"]]),
scr_g_tbl[["PAR short"]] / ( scr_g_tbl[["PAR short"]] + nsc_g_tbl[["PAR short"]])
)
mean(Oki_Osa_scr_ratio_short_arm)## [1] 0.3003286
t.test(Oki_Osa_scr_ratio_long_arm, Oki_Osa_scr_ratio_short_arm, paired = T)##
## Paired t-test
##
## data: Oki_Osa_scr_ratio_long_arm and Oki_Osa_scr_ratio_short_arm
## t = -5.9254, df = 2, p-value = 0.02732
## alternative hypothesis: true mean difference is not equal to 0
## 95 percent confidence interval:
## -0.31610038 -0.05015222
## sample estimates:
## mean difference
## -0.1831263Session information
## R version 4.3.1 (2023-06-16)
## Platform: x86_64-pc-linux-gnu (64-bit)
## Running under: Debian GNU/Linux 12 (bookworm)
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/blas/libblas.so.3.11.0
## LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.11.0
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## time zone: Etc/UTC
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats4 stats graphics grDevices utils datasets methods
## [8] base
##
## other attached packages:
## [1] BSgenome.Oidioi.genoscope.OdB3_1.0.0
## [2] BSgenome.Oidioi.OIST.AOM.5.5f_1.0.1
## [3] BSgenome.Oidioi.OIST.KUM.M3.7f_1.0.1
## [4] BSgenome.Oidioi.OIST.Bar2.p4_1.0.1
## [5] BSgenome.Oidioi.OIST.OSKA2016v1.9_1.0.0
## [6] BSgenome.Oidioi.OIST.OKI2018.I69_1.0.1
## [7] patchwork_1.1.2
## [8] heatmaps_1.24.0
## [9] GenomicFeatures_1.52.1
## [10] AnnotationDbi_1.62.2
## [11] Biobase_2.60.0
## [12] OikScrambling_5.1.0
## [13] ggplot2_3.4.4
## [14] GenomicBreaks_0.14.3
## [15] BSgenome_1.68.0
## [16] rtracklayer_1.60.1
## [17] Biostrings_2.68.1
## [18] XVector_0.40.0
## [19] GenomicRanges_1.52.1
## [20] GenomeInfoDb_1.36.4
## [21] IRanges_2.34.1
## [22] S4Vectors_0.38.2
## [23] BiocGenerics_0.46.0
##
## loaded via a namespace (and not attached):
## [1] splines_4.3.1 BiocIO_1.10.0
## [3] filelock_1.0.2 bitops_1.0-7
## [5] tibble_3.2.1 R.oo_1.25.0
## [7] XML_3.99-0.15 rpart_4.1.19
## [9] rGADEM_2.48.0 lifecycle_1.0.3
## [11] rprojroot_2.0.4 lattice_0.20-45
## [13] MASS_7.3-58.2 backports_1.4.1
## [15] magrittr_2.0.3 Hmisc_5.1-1
## [17] sass_0.4.7 rmarkdown_2.25
## [19] jquerylib_0.1.4 yaml_2.3.7
## [21] plotrix_3.8-3 DBI_1.1.3
## [23] CNEr_1.36.0 minqa_1.2.6
## [25] RColorBrewer_1.1-3 ade4_1.7-22
## [27] abind_1.4-5 zlibbioc_1.46.0
## [29] purrr_1.0.2 R.utils_2.12.2
## [31] RCurl_1.98-1.13 nnet_7.3-18
## [33] pracma_2.4.2 rappdirs_0.3.3
## [35] GenomeInfoDbData_1.2.10 seqLogo_1.66.0
## [37] gdata_3.0.0 annotate_1.78.0
## [39] pkgdown_2.0.7 codetools_0.2-19
## [41] DelayedArray_0.26.7 xml2_1.3.5
## [43] tidyselect_1.2.0 shape_1.4.6
## [45] ggseqlogo_0.1 farver_2.1.1
## [47] lme4_1.1-35.1 BiocFileCache_2.8.0
## [49] matrixStats_1.0.0 base64enc_0.1-3
## [51] GenomicAlignments_1.36.0 jsonlite_1.8.7
## [53] mitml_0.4-5 Formula_1.2-5
## [55] survival_3.5-3 iterators_1.0.14
## [57] systemfonts_1.0.5 foreach_1.5.2
## [59] progress_1.2.2 tools_4.3.1
## [61] ragg_1.2.5 Rcpp_1.0.11
## [63] glue_1.6.2 gridExtra_2.3
## [65] pan_1.9 xfun_0.41
## [67] MatrixGenerics_1.12.3 EBImage_4.42.0
## [69] dplyr_1.1.3 withr_2.5.2
## [71] fastmap_1.1.1 boot_1.3-28.1
## [73] fansi_1.0.5 digest_0.6.33
## [75] R6_2.5.1 mice_3.16.0
## [77] textshaping_0.3.7 colorspace_2.1-0
## [79] GO.db_3.17.0 gtools_3.9.4
## [81] poweRlaw_0.70.6 jpeg_0.1-10
## [83] biomaRt_2.56.1 RSQLite_2.3.3
## [85] weights_1.0.4 R.methodsS3_1.8.2
## [87] utf8_1.2.4 tidyr_1.3.0
## [89] generics_0.1.3 data.table_1.14.8
## [91] prettyunits_1.2.0 httr_1.4.7
## [93] htmlwidgets_1.6.2 S4Arrays_1.0.6
## [95] pkgconfig_2.0.3 gtable_0.3.4
## [97] blob_1.2.4 htmltools_0.5.7
## [99] fftwtools_0.9-11 scales_1.2.1
## [101] png_0.1-8 knitr_1.45
## [103] rstudioapi_0.15.0 tzdb_0.4.0
## [105] reshape2_1.4.4 rjson_0.2.21
## [107] curl_5.1.0 checkmate_2.3.0
## [109] nlme_3.1-162 nloptr_2.0.3
## [111] cachem_1.0.8 stringr_1.5.0
## [113] KernSmooth_2.23-20 parallel_4.3.1
## [115] genoPlotR_0.8.11 foreign_0.8-84
## [117] restfulr_0.0.15 desc_1.4.2
## [119] pillar_1.9.0 grid_4.3.1
## [121] vctrs_0.6.4 jomo_2.7-6
## [123] dbplyr_2.3.3 xtable_1.8-4
## [125] cluster_2.1.4 htmlTable_2.4.2
## [127] evaluate_0.23 readr_2.1.4
## [129] cli_3.6.1 locfit_1.5-9.8
## [131] compiler_4.3.1 Rsamtools_2.16.0
## [133] rlang_1.1.2 crayon_1.5.2
## [135] labeling_0.4.3 plyr_1.8.9
## [137] fs_1.6.3 stringi_1.7.12
## [139] BiocParallel_1.34.2 munsell_0.5.0
## [141] tiff_0.1-11 glmnet_4.1-8
## [143] Matrix_1.5-3 hms_1.1.3
## [145] bit64_4.0.5 KEGGREST_1.40.1
## [147] highr_0.10 SummarizedExperiment_1.30.2
## [149] broom_1.0.5 memoise_2.0.1
## [151] bslib_0.5.1 bit_4.0.5