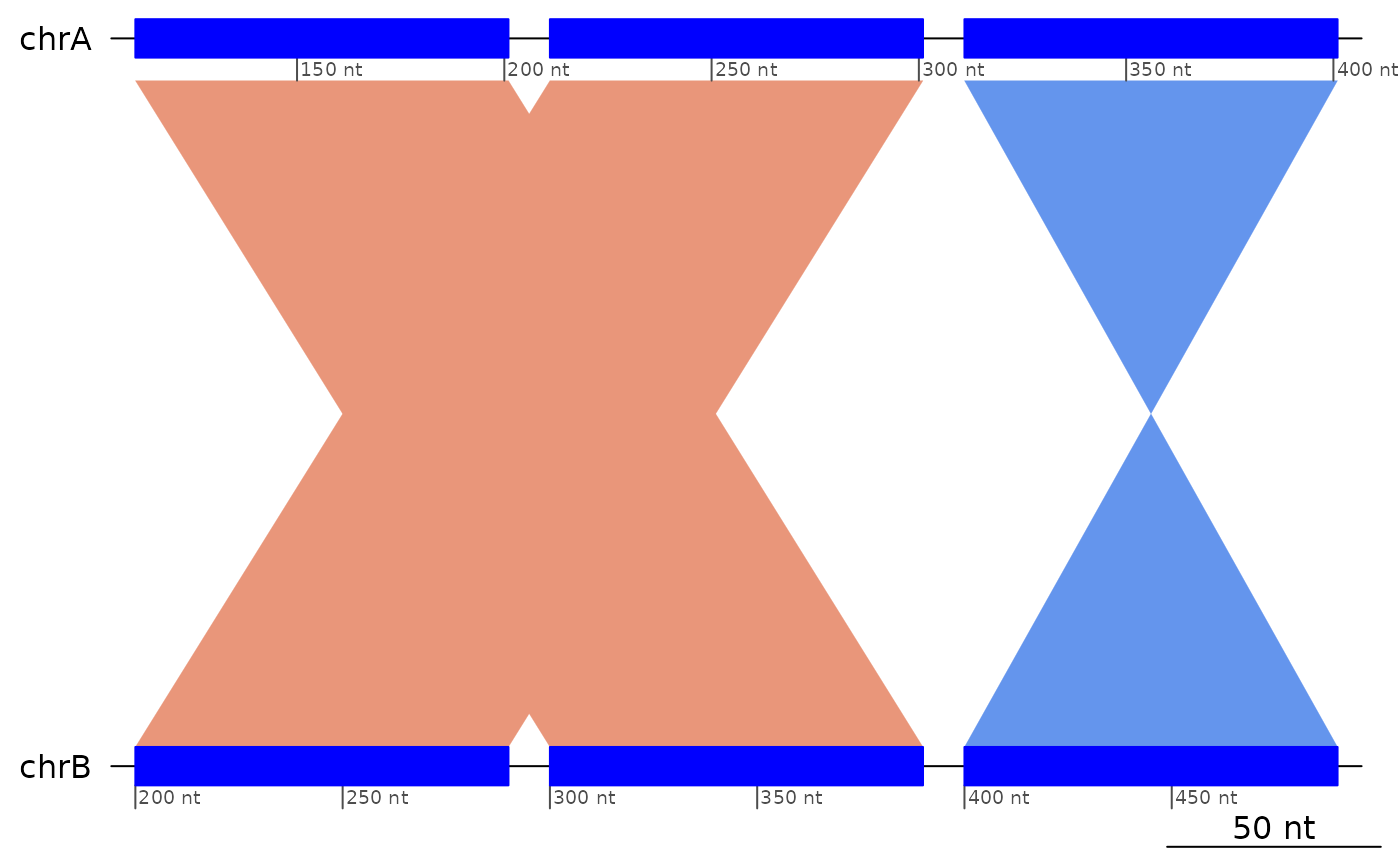
Two consecutive and overlapping inversions will generate patterns that can this function aims to detect.
Arguments
- gb
A
GBreaks()object.- details
Returns more metadata columns if
TRUE.
See also
Other Flagging functions:
flagAll(),
flagColinearAlignments(),
flagInversions(),
flagLongShort(),
flagPairs(),
flagTranslocations(),
flagTwinInversions()
Other Inversion functions:
filterDoubleInversions(),
filterInversions(),
flagInversions(),
flagPairs(),
flagTwinInversions(),
flipInversions(),
leftInversionGaps(),
removeDoubleInversions(),
removeInversions(),
showInversions()
Other Structural variants:
StructuralVariants,
flagInversions(),
flagPairs(),
flagTranslocations(),
flagTwinInversions(),
plotApairOfChrs()
Examples
# Start colinear. Lower case meands minus strand
z0 <- GBreaks(target = GRanges(c(A="T:10-15:+", B="T:20-25:+", C="T:30-35:+")),
query = GRanges(c(A="Q:10-15", B="Q:20-25", C="Q:30-35")))
# Swap coordinates of A and B on the query and flip strands ABC -> baC
z1 <- GBreaks(target = GRanges(c(A="T:10-15:-", B="T:20-25:-", C="T:30-35:+")),
query = GRanges(c(a="Q:20-25", b="Q:10-15", C="Q:30-35")))
# Now query order is b - a - C. Swap a and C and flip strands baC -> bcA
z2 <- GBreaks(target = GRanges(c(A="T:10-15:+", B="T:20-25:-", C="T:30-35:-")),
query = GRanges(c(A="Q:30-35", b="Q:10-15", c="Q:20-25")))
# Altogether, there are:
# ABC -> baC -> bcA
# ABC -> Acb -> Cab
# cba -> BCa -> BAc
# cba -> cAB -> aCB
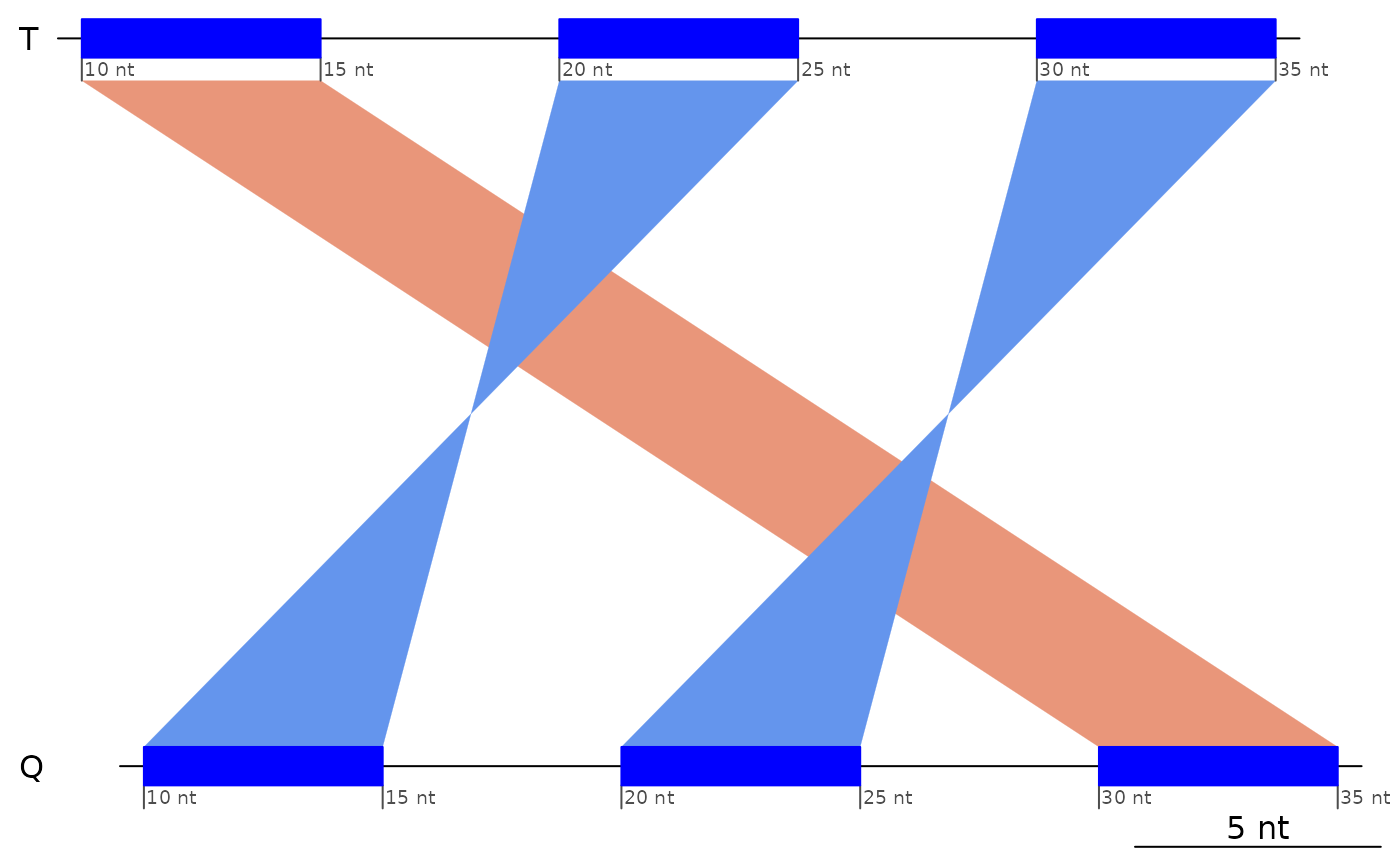
# z2 has same topoloty as package example
plotApairOfChrs(z2)
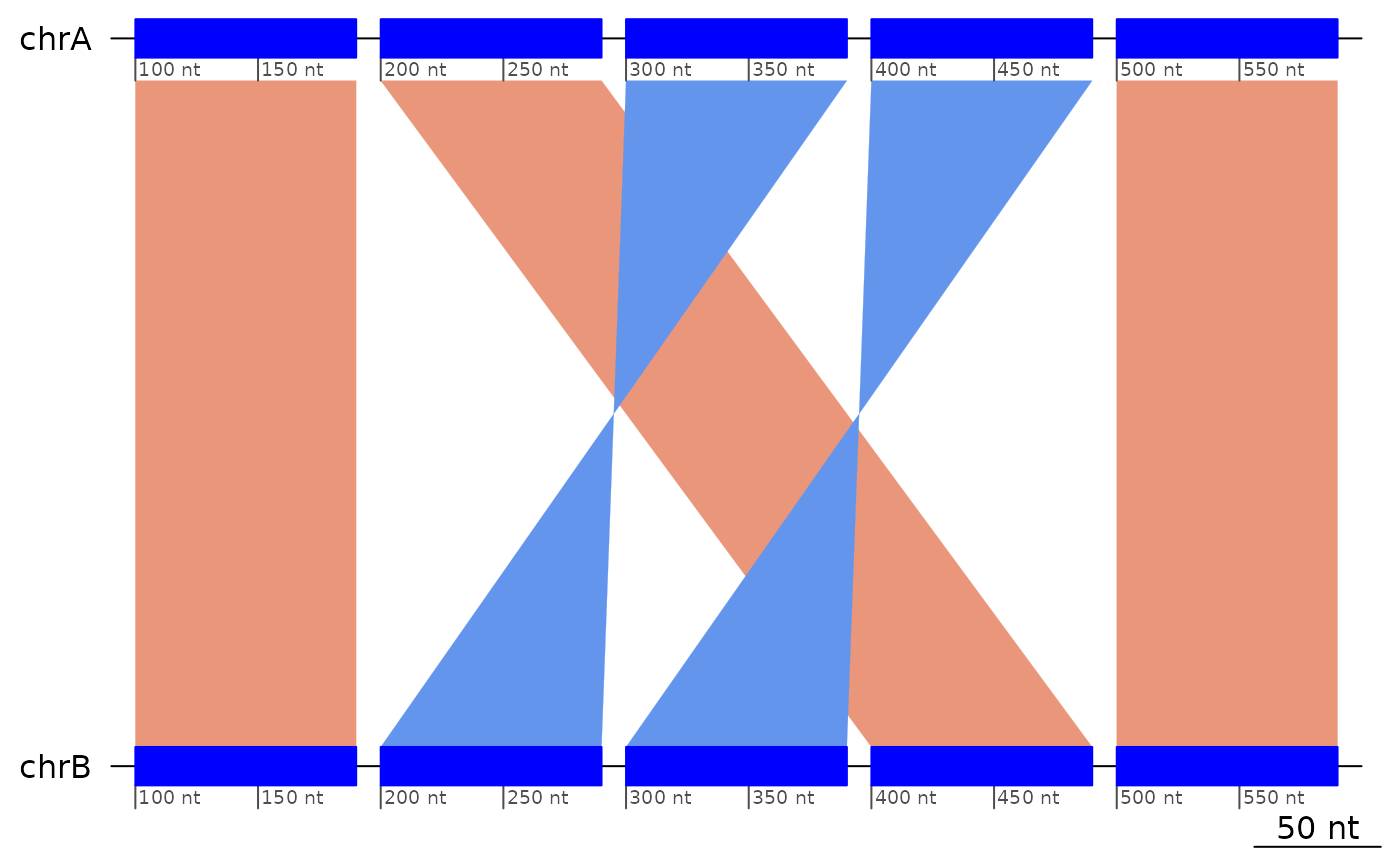
 plotApairOfChrs(exampleDoubleInversion1)
plotApairOfChrs(exampleDoubleInversion1)
 flagDoubleInversions(exampleDoubleInversion1 )
#> GBreaks object with 5 ranges and 2 metadata columns:
#> seqnames ranges strand | query Dbl
#> <Rle> <IRanges> <Rle> | <GRanges> <logical>
#> [1] chrA 100-190 + | chrB:100-190 FALSE
#> [2] chrA 200-290 + | chrB:400-490 TRUE
#> [3] chrA 300-390 - | chrB:200-290 FALSE
#> [4] chrA 400-490 - | chrB:300-390 FALSE
#> [5] chrA 500-590 + | chrB:500-590 FALSE
#> -------
#> seqinfo: 1 sequence from an unspecified genome
exampleDoubleInversion1Rev <- reverse(exampleDoubleInversion1) |> sort(ignore.strand = TRUE)
exampleDoubleInversion1Rev[2:4] |> plotApairOfChrs()
flagDoubleInversions(exampleDoubleInversion1 )
#> GBreaks object with 5 ranges and 2 metadata columns:
#> seqnames ranges strand | query Dbl
#> <Rle> <IRanges> <Rle> | <GRanges> <logical>
#> [1] chrA 100-190 + | chrB:100-190 FALSE
#> [2] chrA 200-290 + | chrB:400-490 TRUE
#> [3] chrA 300-390 - | chrB:200-290 FALSE
#> [4] chrA 400-490 - | chrB:300-390 FALSE
#> [5] chrA 500-590 + | chrB:500-590 FALSE
#> -------
#> seqinfo: 1 sequence from an unspecified genome
exampleDoubleInversion1Rev <- reverse(exampleDoubleInversion1) |> sort(ignore.strand = TRUE)
exampleDoubleInversion1Rev[2:4] |> plotApairOfChrs()